Trans-Splicing Group I Introns: From RNA Repair to Programmable Biocomputing
This article explores the transformative potential of trans-splicing group I intron ribozymes as powerful tools for synthetic biology and biocomputing.
Trans-Splicing Group I Introns: From RNA Repair to Programmable Biocomputing
Abstract
This article explores the transformative potential of trans-splicing group I intron ribozymes as powerful tools for synthetic biology and biocomputing. We cover foundational mechanisms, from their natural self-splicing and mobility to their engineering into trans-splicing devices. The core of the discussion focuses on cutting-edge methodological applications, including the design of complex genetic circuits for cellular logic computation and therapeutic mRNA repair. We also address critical troubleshooting and optimization strategies for enhancing splicing efficiency and specificity, and provide a comparative validation of ribozymes from different species like Tetrahymena and Azoarcus. This resource is tailored for researchers and drug development professionals seeking to harness RNA-based systems for advanced biomedical applications and sophisticated cellular programming.
The Architectural Blueprint: Understanding Group I Intron Structure and Native Function
Group I introns are a distinct class of large, self-splicing ribozymes—catalytic RNA molecules—that excise themselves from mRNA, tRNA, and rRNA precursors through an autocatalytic process [1] [2]. The landmark discovery of the first group I intron in the ribosomal RNA of Tetrahymena thermophila in the early 1980s fundamentally altered our understanding of RNA's biological role, revealing that RNA could possess enzymatic activity independent of proteins [2] [3]. These genetic elements are characterized by their ability to perform splicing via two sequential transesterification reactions, requiring no spliceosome [1]. Ranging in size from approximately 250 to 500 nucleotides, group I introns are found across the tree of life, present in bacteria, bacteriophages, eukaryotic nuclei, and the organelles of lower eukaryotes and plants [1] [4] [5]. Their sporadic phylogenetic distribution and complex evolutionary history, featuring both vertical inheritance and lateral transfer, make them fascinating subjects for studying RNA evolution and mobility [4] [6].
For biocomputing research, group I introns present a compelling platform as naturally occurring, programmable RNA catalysts. Their ability to be engineered for trans-splicing reactions, where the ribozyme acts on a separate substrate RNA molecule, opens avenues for developing synthetic biological circuits and RNA-based computing devices [7] [3]. The precise, sequence-specific recognition and modification of RNA substrates by engineered group I ribozymes can be harnessed to create logical gates, sensors, and signal amplifiers within living cells, forming the foundation of novel biocomputing systems.
Structural and Mechanistic Hallmarks
Conserved RNA Architecture
The catalytic proficiency of group I introns stems from a highly conserved core tertiary structure, despite significant variation at the primary sequence level [1] [2]. The core secondary structure consists of up to ten paired regions (P1-P10) that fold into two primary domains [1] [5]. The P4-P6 domain (comprising P5, P4, P6, and P6a helices) forms a structural scaffold, while the P3-P9 domain (including P8, P3, P7, and P9 helices) constitutes the catalytic center [1]. Short, conserved sequence elements (P, Q, R, and S) form long-range pairing interactions (P-Q and R-S) that are critical for maintaining the active architecture of the ribozyme core [5].
Table 1: Structural Domains of Group I Intron Ribozymes
| Domain | Structural Elements | Functional Role |
|---|---|---|
| Scaffold Domain (P4-P6) | P5, P4, P6, P6a helices | Provides structural framework and stability |
| Catalytic Domain (P3-P9) | P8, P3, P7, P9 helices | Contains active site for splicing catalysis |
| Substrate Domain | P1 and P10 helices | Recognizes and binds 5' and 3' splice sites |
Based on variations in their secondary structure configurations and peripheral elements, group I introns are classified into five main subgroups (IA, IB, IC, ID, IE), which are further divided into at least 17 specific subtypes [2]. This structural diversity reflects the evolutionary adaptability of the core ribozyme scaffold.
The Splicing Mechanism
Group I intron splicing proceeds via two consecutive transesterification reactions that require no external energy source [1] [3]. The process is initiated when an exogenous guanosine nucleotide (exoG) docks into the G-binding site located in the P7 helix [1] [5]. The 3'-OH group of this guanosine acts as a nucleophile, attacking the phosphodiester bond at the 5' splice site within the P1 helix. This first step results in the exoG becoming covalently attached to the 5' end of the intron and liberates the upstream exon with a free 3'-OH group [3].
For the second step, the terminal guanosine of the intron (ωG) displaces the exoG and occupies the G-binding site. The free 3'-OH of the upstream exon then attacks the phosphodiester bond at the 3' splice site (defined by the P10 helix), leading to exon ligation and release of the linear intron RNA [1] [5]. The reaction is catalyzed by a two-metal-ion mechanism similar to that used by protein polymerases and phosphatases, with magnesium ions playing critical roles in stabilizing transition states and activating nucleophiles [1] [5].
Figure 1: The Two-Step Transesterification Mechanism of Group I Intron Splicing. The process is initiated by an exogenous guanosine (exoG) and results in precise exon ligation and intron excision.
Distribution and Evolutionary Dynamics
Phylogenetic Distribution
Group I introns display a widespread but highly sporadic distribution across the tree of life [4]. In bacteria, they are found in tRNA, rRNA, and occasionally protein-coding genes, though their occurrence appears more limited compared to lower eukaryotes [1] [5]. Bacterial group I introns are particularly prevalent in cyanobacteria and Gram-positive bacteria, and they are also found in various bacteriophages that infect these organisms [1]. In eukaryotic microorganisms, including fungi, algae, and protists, group I introns frequently interrupt nuclear rRNA genes as well as mitochondrial and chloroplast genes [2] [4]. The nuclear rDNA of myxomycetes (plasmodial slime molds) represents an especially rich reservoir of diverse group I introns, with some species like Diderma niveum harboring more than 20 introns within a single rRNA primary transcript [3].
Notably, group I introns are absent from bilateral metazoans, with rare exceptions in a few non-bilateral basal lineages and five shark species [2]. This patchy distribution reflects a complex evolutionary history involving both vertical inheritance and extensive horizontal transfer [4] [6].
Table 2: Distribution of Group I Introns Across Biological Kingdoms
| Organism Group | Common Genomic Locations | Prevalence |
|---|---|---|
| Bacteria & Bacteriophages | rRNA, tRNA, phage protein-coding genes | Sporadic but widespread |
| Fungi | Nuclear rDNA, mitochondrial genes | Very common in some lineages |
| Algae & Plants | Chloroplast & mitochondrial genomes | Frequent in organelles |
| Myxomycetes | Nuclear ribosomal DNA | Exceptionally abundant |
| Metazoans | - | Largely absent |
Mobility Mechanisms and Evolutionary Trajectories
Group I introns employ sophisticated mobility mechanisms that enable their spread within and between genomes. Approximately one-fourth to one-third of group I introns contain open reading frames (ORFs) that encode homing endonucleases (HEs) [2]. These highly specific DNA endonucleases initiate a process called "homing" by recognizing and cleaving intronless cognate alleles at specific target sequences [4] [5]. The subsequent DNA repair process using the intron-containing allele as a template results in the conversion of the intronless allele to an intron-containing one, enabling super-Mendelian inheritance of the intron [2].
The evolutionary lifecycle of group I introns and their associated HEs follows a cyclical pattern known as the "homing cycle" [2]. Once an intron becomes fixed in a population through homing, selective pressure to maintain a functional HE diminishes, leading to its degeneration through genetic drift. Eventually, the intron itself may be lost from the population, allowing empty alleles to re-emerge and potentially be invaded again, completing the cycle [2]. Some HEs escape this degenerative fate by acquiring maturase activity, wherein they assist in the folding and splicing of their host intron [2] [4]. This bifunctionality creates a selective advantage for maintaining the HE, as it becomes essential for proper gene expression of the host organism.
An alternative mobility pathway, particularly for introns lacking HEs, is reverse splicing [6]. In this RNA-mediated process, a free intron RNA can reinsert itself into a homologous or heterologous RNA transcript through the reverse of the splicing reaction. Subsequent reverse transcription and recombination can then genomic the insertion. Reverse splicing may explain the long-distance movement of group I introns to non-homologous sites and their spread between evolutionarily distant taxa [6].
Application Notes for Biocomputing Research
Engineering Trans-Splicing Ribozymes
The conversion of natural cis-splicing group I introns into trans-splicing configurations provides a powerful platform for programming RNA-based computational operations in biological systems [7] [3]. In trans-splicing ribozymes, the 5' exon is removed, and the ribozyme's 5' terminus is redesigned to base-pair with a complementary target site on a separate substrate RNA molecule. Upon binding, the ribozyme catalyzes a splicing reaction that replaces the 3' portion of the substrate RNA with the 3' exon carried by the ribozyme [7].
This precise RNA reprogramming capability can be harnessed for multiple biocomputing applications:
- Logic Gate Implementation: Engineered ribozymes can function as Boolean logic gates by designing their activity to be conditional on the presence of specific input RNAs (e.g., AND, OR, NOT gates) [3].
- Signal Amplification Cascades: The catalytic nature of ribozymes enables them to process multiple substrate molecules, allowing for the construction of signal amplification pathways within synthetic genetic circuits.
- State Memory Devices: Trans-splicing events that create stable, heritable changes in RNA or subsequent protein expression can serve as biological memory elements in cellular computing systems.
Comparative Analysis of Model Ribozyme Systems
The selection of appropriate group I intron scaffolds is critical for optimizing the performance of engineered ribozymes in biocomputing applications. Two particularly well-characterized systems offer complementary advantages:
Tetrahymena thermophila Ribozyme (Subgroup IC1): This 414-nucleotide ribozyme represents the historical prototype for group I intron studies [7] [3]. Its extensive characterization provides a rich knowledge base for engineering, though its relatively large size and complex folding pathway may present challenges for some applications [7].
Azoarcus Bacterial Ribozyme (Subgroup IC3): At only 205 nucleotides, the Azoarcus ribozyme is approximately half the size of the Tetrahymena ribozyme and exhibits significantly faster folding kinetics in vitro [7]. Its compact architecture and efficient catalysis make it an attractive candidate for engineering minimal computing elements, though its trans-splicing efficiency in cellular environments requires further optimization [7].
Table 3: Comparison of Model Group I Introns for Biocomputing Applications
| Characteristic | Tetrahymena thermophila | Azoarcus sp. |
|---|---|---|
| Subgroup Classification | IC1 | IC3 |
| Length | ~414 nucleotides | ~205 nucleotides |
| Natural Origin | Nuclear LSU rRNA gene | Bacterial tRNA-Ile gene |
| Folding Kinetics | Slter, more complex | Faster, more efficient |
| Structural Characterization | Extensive | High-resolution crystal structures |
| Trans-Splicing Efficiency | Moderate in cells | High in vitro, lower in cells |
Experimental Protocol: In Vitro Trans-Splicing Assay
This protocol describes a standardized method for assessing the activity of engineered group I intron ribozymes in trans-splicing reactions under near-physiological conditions in vitro [7].
Materials and Reagents
- Purified Ribozyme RNA: In vitro transcribed and gel-purified
- Substrate RNA: Containing target sequence for ribozyme recognition
- Reaction Buffer: 50 mM HEPES (pH 7.5), 100 mM NaCl, 5-10 mM MgCl₂
- NTPs: 1 mM GTP (initiates splicing), 0.5 mM ATP, CTP, UTP
- Stop Solution: 90% formamide, 50 mM EDTA
- Polyacrylamide Gel Equipment: For denaturing urea-PAGE analysis
Procedure
- Ribozyme Annealing: Denature the ribozyme RNA (1-5 µM) at 95°C for 1 minute in reaction buffer without MgCl₂, then cool slowly to 37°C over 15 minutes to promote proper folding.
- Reaction Initiation: Add MgCl₂ to a final concentration of 5-10 mM along with substrate RNA (1-5 µM) and GTP (1 mM). Mix thoroughly and incubate at 37°C.
- Timepoint Sampling: Remove aliquots at predetermined timepoints (e.g., 0, 5, 15, 30, 60, 120 minutes) and mix immediately with stop solution to quench the reaction.
- Product Analysis: Resolve the reaction products by denaturing urea-PAGE (6-10% acrylamide). Visualize RNA bands using ethidium bromide, SYBR Gold, or radioisotopic labeling.
- Quantification: Determine the percentage of spliced product relative to total substrate using densitometry or phosphorimaging analysis.
Optimization Notes
- The Extended Guide Sequence (EGS) design should be optimized for each ribozyme-substrate pair. For Azoarcus ribozymes, designs that mimic the natural anticodon stem-loop context of the native tRNA environment often yield superior activity [7].
- Magnesium concentration (typically 5-10 mM) and temperature (37-45°C) should be systematically optimized for each engineered ribozyme.
- For kinetic analysis, multiple substrate concentrations should be tested to determine Michaelis-Menten parameters (Kₘ and kcat).
Figure 2: Experimental Workflow for Assessing Trans-Splicing Ribozyme Activity In Vitro.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for Group I Intron and Biocomputing Applications
| Reagent/Category | Specifications | Research Application |
|---|---|---|
| In Vitro Transcription Kits | T7/SP6 RNA polymerase-based systems | Production of catalytic RNA components |
| RNA Purification Materials | Denaturing PAGE systems or FPLC | Isolation of highly active ribozyme RNA |
| Magnesium Salts | High-purity MgCl₂ (5-10 mM range) | Essential cofactor for ribozyme folding and catalysis |
| Guanosine Nucleotides | GTP, GMP, or GDP (1 mM typical) | Initiates splicing as exogenous nucleophile |
| Extended Guide Sequences | Custom-designed oligonucleotides (4-20 nt) | Enhances target recognition and binding specificity |
| Fluorescent Reporters | FRET pairs (e.g., Cy3/Cy5) or GFP variants | Real-time monitoring of splicing activity in vitro and in vivo |
| Homing Endonucleases | LAGLIDADG or GIY-YIG family proteins | DNA-level programming of genetic circuits |
Future Perspectives in Biocomputing
The unique catalytic properties of group I introns position them as versatile components for next-generation biocomputing systems. Future research directions should focus on enhancing the predictability and orthogonality of engineered ribozymes to enable the construction of more complex computational networks in cellular environments. Key challenges include improving the in vivo stability and kinetics of compact ribozymes like the Azoarcus system, developing allosteric control mechanisms that regulate ribozyme activity in response to specific molecular inputs, and creating computational models that accurately predict ribozyme-substrate interactions in the context of cellular RNA folding landscapes [7] [3].
The integration of group I intron-based RNA computation with other synthetic biology components—such as CRISPR systems, protein-based logic gates, and cell-free expression platforms—will enable the development of sophisticated hybrid computational devices capable of processing complex biological information for therapeutic, diagnostic, and environmental applications. As our understanding of RNA structure-function relationships deepens, the programmability of group I intron ribozymes will continue to expand, solidifying their role as fundamental components in the emerging toolkit of biological computing.
The two-step transesterification splicing pathway is the fundamental chemical mechanism used by group I intron ribozymes to self-excise from RNA transcripts and ligate the flanking exons. This process is catalyzed entirely by the catalytic RNA core of the intron, without the requirement for protein enzymes, making it a cornerstone mechanism for biocomputing research and synthetic biology applications. The pathway relies on consecutive phosphoester transfers that rearrange the RNA backbone, resulting in precise splicing outcomes. Understanding this core mechanism enables researchers to engineer trans-splicing group I introns for diverse applications including RNA repair, therapeutic development, and molecular programming [3] [8].
For synthetic biologists and drug development professionals, this self-splicing mechanism offers a programmable RNA processing system with predictable kinetics and modular components. The ribozyme's ability to function in trans—splicing together exons from separate RNA molecules—enables innovative approaches for rewiring genetic circuits and developing RNA-based therapeutics. Recent advances have demonstrated the clinical potential of this technology, with FDA-approved Phase I/IIa IND trials for trans-splicing ribozymes in cancer treatment [8].
Detailed Mechanism of Two-Step Transesterification
Biochemical Pathway
The two-step transesterification mechanism proceeds through defined sequential reactions that require specific cofactors and produce characteristic intermediate structures:
Step 1: 5' Splice Site Cleavage - The reaction is initiated when the 3' hydroxyl group (3'OH) of an exogenous guanosine cofactor (exoG, typically GTP) performs a nucleophilic attack on the phosphodiester bond at the 5' splice site. This transesterification reaction results in cleavage at the 5' splice site and covalent attachment of the guanosine to the 5' end of the intron RNA. The upstream exon is released with a free 3'OH group, while the guanosine cofactor becomes attached to the intron's 5' terminus [3] [9].
Step 2: 3' Splice Site Cleavage and Exon Ligation - The 3'OH group of the released 5' exon now acts as a nucleophile, attacking the phosphodiester bond at the 3' splice site. This second transesterification results in ligation of the flanking exons and release of the intron RNA. The intron is excised as a linear molecule containing the initially added guanosine at its 5' end [3] [10].
Table 1: Key Components of the Transesterification Reaction
| Component | Role in Mechanism | Chemical Function |
|---|---|---|
| Exogenous Guanosine (exoG) | Nucleophile initiator | Provides free 3'OH for first nucleophilic attack |
| 5' Splice Site | First reaction site | Phosphate bond attacked by exoG 3'OH |
| 3' Splice Site | Second reaction site | Phosphate bond attacked by exon 3'OH |
| ωG (Omega G) | Terminal intron nucleotide | Participates in guanosine binding site in catalytic core |
| Catalytic RNA Core | Reaction catalyst | Precisely positions substrates for transesterification |
Structural Requirements
The group I intron ribozyme folds into a conserved tertiary structure with specific domains essential for catalysis. The catalytic core consists of paired RNA segments (P3-P9) organized into three structural domains: the substrate domain (P1-P2), scaffold domain (P4-P6), and catalytic domain (P3-P7). The P7 helix contains the guanosine binding site (G site) where the exogenous guanosine cofactor initially binds before the first transesterification step. The internal guide sequence within P1 facilitates specific recognition of the 5' splice site through base-pairing interactions [3].
The reaction requires magnesium ions (Mg²⁺) in the catalytic core, which serve to stabilize the transition state and facilitate the transesterification chemistry. The same catalytic mechanism involving two magnesium ions is employed by the spliceosome, suggesting an evolutionary relationship between self-splicing ribozymes and the eukaryotic splicing machinery [10] [3].
Experimental Protocols and Applications
In Vitro Splicing Assay Protocol
Objective: To demonstrate and analyze group I intron self-splicing via the two-step transesterification pathway in vitro.
Materials:
- DNA template encoding the group I intron flanked by exons
- T7 or SP6 RNA polymerase for in vitro transcription
- Nucleotide triphosphates (NTPs)
- Guanosine triphosphate (GTP)
- Magnesium chloride (MgCl₂)
- Buffer components (Tris-HCl, pH 7.5)
- Stop solution (EDTA, formamide)
- Polyacrylamide gel electrophoresis equipment
Method:
- Template Preparation: Linearize plasmid DNA containing the group I intron construct or prepare by PCR amplification. For the CIRC method, ensure the construct contains an intact group I intron [9].
In Vitro Transcription: Synthesize the precursor RNA using T7 RNA polymerase in a reaction containing:
- 1 μg DNA template
- 2 mM each NTP
- 40 mM Tris-HCl, pH 7.5
- 10 mM MgCl₂
- 5 mM DTT
- 2 mM spermidine
- Incubate at 37°C for 2 hours [9]
Splicing Reaction: Set up the splicing reaction with:
- Precursor RNA (0.1-1.0 μM)
- 40 mM Tris-HCl, pH 7.5
- 10-100 mM MgCl₂ (concentration affects efficiency)
- 1 mM GTP
- Incubate at 37°C for 30-60 minutes [9]
Reaction Termination: Add EDTA to 25 mM final concentration to chelate Mg²⁺ and stop the reaction.
Product Analysis:
- Separate RNA products by denaturing polyacrylamide gel electrophoresis (8% PAGE, 7M urea)
- Visualize using ethidium bromide, SYBR Gold, or autoradiography if using radiolabeled RNA
- Identify bands corresponding to precursor RNA, spliced exons, and excised intron
Troubleshooting Notes:
- For introns with non-G 5' termini, additional 5' G residues may be needed for efficient T7 transcription without affecting circularization efficiency [9].
- Mg²⁺ concentration optimization is critical—test range from 10-100 mM
- Time course experiments (0-120 minutes) can reveal splicing kinetics
Trans-Splicing Applications for RNA Repair
The two-step transesterification mechanism can be harnessed for therapeutic RNA repair using engineered group I intron ribozymes that operate in trans. This approach enables correction of disease-causing mutations at the RNA level:
Protocol for Targeted RNA Repair:
Ribozyme Design:
- Engineer the Tetrahymena thermophila group I intron to recognize a specific target mRNA
- Modify the Internal Guide Sequence (IGS) to base-pair with the target RNA upstream of the mutation site
- Include the correct sequence in the ribozyme's 3' exon to replace the mutated region [8]
Splice Site Identification:
- Computationally predict accessible uridine residues in the target RNA using free energy calculations (IntaRNA2 software)
- Select splice sites with favorable binding energies (ΔG) for ribozyme recognition [8]
Efficiency Optimization:
- Incorporate an Extended Guide Sequence (EGS) with 3 nucleotides forming a P1 extension (P1ex)
- Include an antisense duplex (8-46 bp) for enhanced target binding
- Test EGS libraries with randomized internal loops to identify high-efficiency variants [8]
Validation in Cellular Models:
- Transfert engineered ribozyme into target cells (e.g., HEK293 NF1-/- for neurofibromatosis type I model)
- Analyze trans-splicing products by RT-PCR and sequencing
- Confirm functional protein expression via Western blot or functional assays [8]
Table 2: Quantitative Comparison of Splicing Methods
| Method | Splicing Efficiency | Mg²⁺ Requirement | Incubation Time | Key Applications |
|---|---|---|---|---|
| CIRC (Complete Intron) | High | Low (mild conditions) | Short | Large RNA circularization (>12 kb) |
| PIE (Permuted Intron-Exon) | Moderate | High (high Mg²⁺) | Extended | Standard circRNA production |
| PIET (Trans-Splicing) | Moderate to High | Adjustable | Controlled by component addition | Regulated splicing applications |
| Therapeutic Trans-Splicing | Variable (enhanceable with EGS) | Physiological | Dependent on delivery | NF1, cancer, genetic disorders |
Visualization of Splicing Pathways
Two-Step Transesterification Mechanism
RNA Circularization Methods Comparison
Research Reagent Solutions
Table 3: Essential Research Reagents for Trans-Splicing Experiments
| Reagent/Category | Specific Examples | Function in Research | Protocol Notes |
|---|---|---|---|
| Group I Intron Sources | Tetrahymena thermophila, Anabaena (Ana) | Catalytic RNA core for splicing | CIRC method uses intact forms [9] |
| RNA Purification Tools | RNase R, Oligo(dT) beads | Circular RNA purification | RNase R degrades linear RNAs only [9] |
| Splicing Cofactors | GTP (Guanosine Triphosphate) | Initiates first transesterification | Not required for CIRC method [9] |
| Magnesium Salts | MgCl₂ | Catalytic ion for ribozyme function | Concentration affects efficiency [9] |
| Target RNA Templates | NF1 mRNA, Dystrophin mRNA | Therapeutic splicing targets | Full-length dystrophin (~12 kb) demonstrated [9] [8] |
| Computational Tools | IntaRNA2 software | Splice site prediction | Calculates binding free energies [8] |
| Delivery Systems | Transfection reagents, Viral vectors | Cellular ribozyme delivery | Critical for therapeutic applications [8] |
| Detection Methods | RT-PCR, RNase R assay | Splicing product validation | Confirms precise exon ligation [9] |
Technical Considerations and Optimization
Enhancing Splicing Efficiency
Multiple strategies can optimize the efficiency of the two-step transesterification pathway for research and therapeutic applications:
Extended Guide Sequences (EGS): Incorporating EGS elements with optimal internal loop configurations can enhance trans-splicing efficiency by over 50-fold. Combinatorial libraries with randomized EGS sequences can identify high-performance variants through barcode selection [8].
Magnesium Optimization: The CIRC method demonstrates enhanced RNA circularization efficiency under mild conditions (lower Mg²⁺ concentrations), preserving RNA integrity while maintaining high splicing yields. Titrate Mg²⁺ between 10-100 mM for optimal results in specific applications [9].
Sequence Engineering: For the CIRC method, removing homology arms required in traditional PIE approaches significantly enhances circularization efficiency. Additionally, 5' terminal G residues can be added to facilitate T7 transcription without compromising circularization efficiency [9].
Applications in Biocomputing
The predictable nature of the two-step transesterification mechanism enables its use in synthetic biology and molecular programming:
Logic Gate Construction: Engineered ribozymes can function as programmable RNA processors, executing Boolean operations through controlled splicing events.
Molecular Sensors: Splicing-based biosensors can detect specific RNA sequences through IGS complementarity, triggering detectable output signals via trans-splicing.
RNA Circuitry: Multiple ribozymes can be networked to create complex computational RNA devices that process genetic information and execute programmed responses.
The continued refinement of group I intron trans-splicing technology, particularly through methods like CIRC that offer improved efficiency and simplified implementation, positions this mechanism as a powerful tool for both therapeutic development and biocomputing research.
Group I introns are catalytic RNAs (ribozymes) that excise themselves from primary RNA transcripts and ligate the flanking exons via two transesterification reactions [7]. These natural cis-splicing ribozymes can be engineered into trans-splicing variants capable of modifying separate substrate RNAs, making them powerful tools for biocomputing research and potential therapeutic applications, such as repairing mutated mRNAs [7]. The structural diversity of group I introns is classified into several major subgroups (IA, IB, IC, ID, IE, etc.), which exhibit distinct structural features and biochemical properties [11]. Understanding this classification is paramount for selecting the appropriate ribozyme for specific biocomputing tasks, as characteristics like size, folding kinetics, and optimal splice site recognition vary between subgroups [7] [11]. For instance, the well-characterized Tetrahymena thermophila ribozyme (subgroup IC1) and the smaller, fast-folding Azoarcus ribozyme (subgroup IC3) serve as contrasting models for developing synthetic genetic circuits and RNA-based sensors [7].
Classification and Characteristics of Major Subgroups
The classification of group I introns into subgroups is based on conserved primary sequences and secondary structure features. The table below summarizes the key characteristics of several major subgroups, highlighting their diverse origins and properties relevant to biocomputing applications.
Table 1: Classification and Key Features of Major Group I Intron Subgroups
| Subgroup | Representative Intron | Structural Features | Size (Nucleotides) | Trans-Splicing Efficiency & Notes |
|---|---|---|---|---|
| IC1 | Tetrahymena thermophila (16S rRNA) | Well-characterized conserved core structure [7]. | ~400 [7] | High efficiency in vitro; widely used as a model system; requires optimized EGS for high trans-splicing activity [7]. |
| IC3 | Azoarcus sp. (tRNAIle) | Compact, highly structured core; fast-folding kinetics [7]. | 205 [7] | Efficient in vitro with a design mimicking its natural cis-splicing context; lower efficiency in E. coli cells compared to IC1 [7]. |
| IE | Didymium iridis | Distinct structural adaptations in the catalytic core. | Information Missing | Capable of trans-splicing; efficiency can be improved with an Extended Guide Sequence (EGS) [7]. |
| I (General) | Twort intron (used in structural studies) | Conserved tertiary structure with P4-P6 and P3-P9 domains [11]. | Information Missing | Binds fungal mtTyrRSs (e.g., CYT-18) via a conserved phosphodiester-backbone recognition mechanism [11]. |
The structural divergence between subgroups is primarily localized to specific regions, such as the group I intron binding surface recognized by protein cofactors. Fungal mitochondrial tyrosyl-tRNA synthetases (mtTyrRSs), like the CYT-18 protein from Neurospora crassa, have evolved a specialized binding surface to stabilize the catalytically active RNA structure of group I introns [11]. This surface includes an N-terminal extension (H0) and small insertions (Ins 1, Ins 2), which show significant variation across different Pezizomycotina fungi (e.g., A. nidulans and C. posadasii), contributing to intron-binding specificity [11].
Experimental Protocols for Trans-Splicing Analysis
In Vitro Trans-Splicing Assay for Splice Site Identification
This protocol identifies accessible splice sites (uridine residues) on a target mRNA for a given trans-splicing ribozyme, adapted from studies on the Azoarcus and Tetrahymena ribozymes [7].
- Principle: A pool of ribozymes with a randomized substrate recognition sequence is used. Following the reaction, reverse transcription-PCR (RT-PCR) specifically amplifies the trans-splicing products, which are then sequenced to identify the utilized splice sites [7].
- Materials:
- Purified Ribozyme Pool: T7-transcribed ribozyme with a randomized 9-12 nucleotide Internal Guide Sequence (IGS) at its 5' end.
- Target Substrate RNA: In vitro transcribed model mRNA (e.g., Chloramphenicol acetyl transferase (CAT) mRNA).
- Reaction Buffer: 50 mM HEPES (pH 7.5), 100 mM KCl, 5-10 mM MgCl₂.
- Guanosine Nucleotide: 1 mM GTP or G, to initiate the splicing reaction.
- RT-PCR Kit: Enzymes and reagents for reverse transcription and polymerase chain reaction.
- Procedure:
- Incubation: Mix 1 pmol of the ribozyme pool with 0.5 pmol of the target substrate RNA in reaction buffer. Add 1 mM GTP to initiate the reaction.
- Incubation Conditions: Incubate at 37°C for 60 minutes under near-physiological conditions to mimic the cellular environment.
- RNA Extraction: Post-incubation, purify the RNA products using phenol-chloroform extraction and ethanol precipitation.
- Reverse Transcription (RT): Use a gene-specific primer complementary to the ribozyme's 3' exon to transcribe the RNA products into cDNA.
- PCR Amplification: Amplify the cDNA using primers specific to the 3' exon of the ribozyme and the 5' region of the target substrate.
- Product Analysis: Clone the PCR products and sequence individual clones, or use high-throughput sequencing to identify the sequence at the exon-exon junction, thereby revealing the splice site (U) used on the substrate mRNA.
Protocol for Assessing Trans-Splicing Efficiency with an EGS
This protocol measures the efficiency of a trans-splicing reaction, comparing designs with and without an Extended Guide Sequence (EGS), which provides additional base-pairing to the substrate [7].
- Principle: The ribozyme and substrate are incubated under defined conditions. The products are separated by denaturing polyacrylamide gel electrophoresis (PAGE), and the ratio of spliced product to unreacted substrate is quantified to determine efficiency [7].
- Materials:
- Radiolabeled Substrate RNA: Target RNA body-labeled with ³²P during in vitro transcription.
- Defined Ribozyme: Purified, unlabeled trans-splicing ribozyme, with or without an EGS.
- Stop Solution: 95% formamide, 10 mM EDTA, 0.1% bromophenol blue.
- Procedure:
- Reaction Setup: Combine radiolabeled substrate with an excess of ribozyme (e.g., 5:1 molar ratio) in reaction buffer with MgCl₂ and GTP.
- Time-Course Sampling: Remove aliquots from the reaction mixture at specific time intervals (e.g., 0, 5, 15, 30, 60 min).
- Reaction Termination: Immediately mix each aliquot with the formamide-based stop solution and heat to 95°C for 2 minutes to denature the RNA and halt the reaction.
- Product Separation: Resolve the reaction products by denaturing PAGE (e.g., 8% polyacrylamide, 8 M urea).
- Visualization & Quantification: Expose the gel to a phosphorimager screen and quantify the bands corresponding to the substrate and the trans-spliced product using image analysis software (e.g., ImageQuant). The splicing efficiency is calculated as the percentage of substrate converted to product over time.
The following workflow diagram illustrates the key steps in the analysis of trans-splicing group I introns:
Research Reagent Solutions
Key reagents and their functions for experimental work with trans-splicing group I introns are summarized below.
Table 2: Essential Research Reagents for Trans-Splicing Group I Intron Experiments
| Research Reagent | Function & Application in Trans-Splicing |
|---|---|
| T7 RNA Polymerase | In vitro transcription of ribozyme and substrate RNAs with high yield [7]. |
| ³²P-UTP (Radiolabeled) | Radioactive labeling of RNA for highly sensitive detection and quantification of splicing products via gel electrophoresis and phosphorimaging [7]. |
| Extended Guide Sequence (EGS) | An elongation of the ribozyme's 5' terminus that provides additional base-pairing with the substrate RNA, increasing target specificity and splicing efficiency [7]. |
| CYT-18 Protein (mtTyrRS) | A fungal mitochondrial tyrosyl-tRNA synthetase that functions as a splicing cofactor by binding and stabilizing the catalytically active structure of group I introns [11]. |
| Cloning Vector (e.g., pUC19) | For the molecular cloning of PCR products from RT-PCR assays, enabling sequencing and identification of splice sites [7]. |
Group I introns are not merely self-splicing RNA elements; they are sophisticated mobile genetic entities whose propagation is engineered by highly specific homing endonucleases (HEs). These "selfish" enzymes facilitate the super-Mendelian inheritance of their host introns through a precise molecular mechanism known as homing [2] [12]. In the context of advancing biocomputing research, understanding and harnessing this mobility is paramount. Homing endonucleases function as molecular programmers, inserting genetic code with remarkable precision through a well-characterized double-strand break (DSB) and repair cycle [13] [14]. This application note details the mechanisms, key reagents, and experimental protocols for leveraging the homing cycle in sophisticated gene network design and therapeutic development, framing these natural systems as programmable tools for synthetic biology.
The Molecular Mechanism of the Homing Cycle
The homing cycle is a gene conversion process that enables the copying of a mobile genetic sequence (e.g., a group I intron) into a cognate allele that lacks it. The process is initiated and driven by the homing endonuclease.
The Core Homing Process
The homing cycle can be broken down into a series of discrete, programmable steps, as illustrated in the diagram below.
- Expression and Cleavage: In a heterologous cell containing one allele with the homing endonuclease gene (HEG+) and one without (HEG-), the HE is expressed. The enzyme then recognizes and introduces a site-specific double-strand break (DSB) in the cognate recognition site of the HEG- allele [13] [12].
- Repair and Gene Conversion: The cellular DNA repair machinery, specifically the homology-directed repair (HDR) pathway, uses the homologous HEG+ allele as a template to repair the break. This process copies the HE gene and its associated intron into the previously empty allele, resulting in two HEG+ alleles [13] [14]. The long recognition sites of HEs (12-40 bp) ensure this process occurs with high specificity and minimal off-target effects, a critical feature for biocomputing applications requiring precise logic operations [13].
Distinguishing Features of Homing Endonucleases
Homing endonucleases are uniquely suited for programming gene conversion compared to conventional restriction enzymes. The key differences are summarized in the table below.
Table 1: Key Characteristics of Homing Endonucleases versus Type II Restriction Enzymes
| Feature | Homing Endonucleases | Type II Restriction Enzymes |
|---|---|---|
| Recognition Site | Long (12-40 bp), often asymmetric [13] [12] | Short (4-8 bp), usually palindromic [12] |
| Sequence Tolerance | Tolerant of some degeneracy [13] [12] | Highly specific; variations abolish activity [12] |
| Phylogenetic Distribution | All domains of life (Archaea, Bacteria, Eukarya) [2] [12] | Primarily Bacteria and Archaea [12] |
| Genomic Context | Introns, inteins, or freestanding [13] [12] | Almost always freestanding [12] |
| Primary Function | Self-propagation (homing) [12] | Host defense (restriction) [12] |
Major Families of Homing Endonucleases
Homing endonucleases are classified into distinct families based on conserved amino acid motifs and their structural folds. Understanding these families is essential for selecting the appropriate enzyme for a given application. The major families and their characteristics are detailed below.
Table 2: Major Structural Families of Homing Endonucleases
| Family | Conserved Motif(s) | Oligomeric State | Prototypical Member | Key Features |
|---|---|---|---|---|
| LAGLIDADG | 1 or 2 LAGLIDADG motifs [13] [12] | Monomer or Homodimer [13] | I-CreI, I-DmoI [13] | Most common family; saddle-shaped structure interacting with DNA major groove [12] |
| GIY-YIG | GIY-YIG motif in N-terminal region [13] [12] | Monomer [13] | I-TevI [13] [12] | Modular structure with separable catalytic and DNA-binding domains [13] |
| HNH | H-N-H consensus sequence [13] [12] | Monomer [13] | I-HmuI [13] [12] | Contains a zinc finger domain; related to His-Cys box family [12] |
| His-Cys Box | ~30 aa region with 2 His, 3 Cys [13] [12] | Homodimer [13] | I-PpoI [13] [12] | Metal ion coordination for catalysis; possibly related to H-N-H family [13] [12] |
The Scientist's Toolkit: Key Research Reagent Solutions
Leveraging the homing cycle for research and development requires a specific set of molecular tools. The following table catalogs essential reagents and their functions.
Table 3: Essential Research Reagents for Homing Endonuclease Work
| Research Reagent | Function/Description | Example/Source |
|---|---|---|
| Custom Engineered HEs | Tailored endonucleases re-engineered from wild-type templates (e.g., LAGLIDADG) to recognize non-native DNA sequences for gene targeting [13]. | I-CreI and I-DmoI derivatives [13] |
| Group I Intron Database | A comprehensive, unified database providing group I intron sequences with precise exon-intron boundaries, subtype information, and putative HEs [15]. | https://github.com/LaraSellesVidal/Group1IntronDatabase [15] |
| Trans-splicing Ribozyme Scaffolds | Engineered group I introns (e.g., from Azoarcus or Tetrahymena) that can be repurposed to perform trans-splicing reactions for targeted RNA repair or reprogramming [7]. | Azoarcus ribozyme (IC3 subgroup) [7] |
| MARC1 Mouse Line | A transgenic mouse line containing multiple dormant homing guide RNA (hgRNA) barcoding elements for lineage tracing studies upon crossing with a Cas9-expressing line [16]. | MARC1 (PB3 and PB7 lines) [16] |
| Homing Site Reporters | Plasmid-based assays with an integrated HE recognition site upstream of a reporter gene (e.g., GFP). Cleavage and repair via HDR using a donor template restores reporter function, quantifying HE activity. | Custom cloning required |
Detailed Experimental Protocols
Protocol 1: Quantifying Homing Endonuclease Activity and SpecificityIn Vitro
This protocol outlines a method for assessing the cleavage efficiency and specificity of a purified homing endonuclease.
I. Materials
- Purified homing endonuclease (e.g., I-CreI or engineered variant)
- Target plasmid DNA (containing the cognate recognition site)
- Off-target control plasmid DNA (containing a closely related sequence)
- Reaction buffer (e.g., 50 mM Tris-HCl, pH 7.5, 10 mM MgCl₂, 100 mM NaCl, 1 mM DTT)
- Agarose gel electrophoresis equipment
- DNA staining dye (e.g., GelRed)
II. Methodology
- Reaction Setup: In a 0.5 mL tube, combine 500 ng of target plasmid DNA with 1 µL of purified HE in a total volume of 20 µL of reaction buffer.
- Incubation: Incubate the reaction at 37°C for 1 hour.
- Controls: Set up parallel reactions with (a) no enzyme (negative control) and (b) off-target control plasmid DNA.
- Reaction Termination: Stop the reaction by adding 2 µL of 10x DNA loading dye or by heat-inactivating the enzyme (e.g., 65°C for 15 min).
- Analysis: Load the entire reaction onto a 1% agarose gel. Run the gel at 100V for 45 minutes, visualize under UV light, and document.
III. Data Interpretation
- High Specificity: Complete cleavage of the target plasmid (evidenced by a shift from supercoiled to linear form) with no observable cleavage of the off-target plasmid.
- Low Specificity/Tolerance: Cleavage of both target and off-target plasmids, indicating tolerance for sequence degeneracy, which may require protein re-engineering for therapeutic applications [13].
Protocol 2: Assessing Gene Correction via Homing in Mammalian Cells
This protocol describes a cell-based assay to measure the efficiency of homing endonuclease-mediated gene correction.
I. Materials
- Mammalian cells (e.g., HEK293)
- Expression plasmid for the homing endonuclease
- "Homing Reporter" plasmid: A construct where the HE recognition site is inserted into a defective GFP gene, disrupting its coding sequence.
- "Donor Template" plasmid: Contains the functional HE gene (or another corrective sequence) flanked by homology arms matching the region around the break.
- Transfection reagent
- Flow cytometer
II. Methodology
- Cell Seeding: Seed 2 x 10^5 cells per well in a 12-well plate 24 hours before transfection.
- Transfection: Co-transfect cells with a 1:1:1 molar ratio of the HE expression plasmid, Homing Reporter plasmid, and Donor Template plasmid.
- Control Transfections: Include controls lacking the HE expression plasmid or the Donor Template.
- Incubation: Incubate cells for 48-72 hours to allow for expression, cleavage, and repair.
- Analysis: Harvest cells and analyze by flow cytometry to quantify the percentage of GFP-positive cells, indicating successful homing and gene correction.
III. Data Interpretation
- The percentage of GFP-positive cells in the complete reaction, minus the background from controls, quantifies the homing efficiency.
- This assay validates the enzyme's functionality in a cellular context and its potential for ex vivo gene therapy of monogenic diseases [13].
The experimental workflow for this protocol is illustrated below.
Applications in Biocomputing and Therapeutic Development
The unique properties of the homing system make it a powerful platform for advanced biological programming.
Ex Vivo Gene Therapy for Monogenic Diseases: Custom-designed HEs can correct defective genes with high specificity and low toxicity. The process involves extracting patient cells, correcting the gene defect ex vivo using HE-mediated HDR, and re-infusing the modified cells [13]. This approach is particularly suited for diseases amenable to stem cell or lymphocyte therapy.
Developmental Lineage Barcoding: The MARC1 mouse system utilizes "homing guide RNAs" (hgRNAs). When crossed with a Cas9-expressing line, these hgRNAs self-target and accumulate diverse, heritable mutations during cell divisions. The combinatorial mutation patterns serve as lineage barcodes, enabling the reconstruction of entire cellular lineage trees [16]. This provides a powerful tool for understanding development, cancer, and regeneration.
RNA Reprogramming with Trans-Splicing Ribozymes: Engineered group I introns can be used for trans-splicing to repair or reprogram target mRNAs. This dual-function modality simultaneously reduces disease-associated gene expression and induces therapeutic gene activity specifically in target cells [17]. A hTERT-targeting ribozyme has progressed to clinical trials for cancer treatment [17].
Logic Gate Operations in Synthetic Gene Circuits: The high specificity of HE-DNA recognition allows for the design of complex logic operations. For example, a synthetic circuit could be designed where a specific output gene is activated only upon the simultaneous correction of two different genomic loci by two distinct HEs, effectively creating a genetically encoded AND gate. This leverages the homing cycle's programmability for sophisticated biocomputing.
Trans-splicing represents a fundamental RNA processing mechanism in which exons from two separate pre-mRNA molecules are joined to form a single chimeric RNA transcript. This process stands in contrast to conventional cis-splicing, where exons within the same pre-mRNA molecule are connected [18]. Initially discovered in trypanosomes during RNA processing for variant surface glycoprotein, trans-splicing has since been documented across diverse eukaryotic lineages, from lower eukaryotes to vertebrates [18] [19]. The evolutionary trajectory of trans-splicing reveals dynamic changes across species, with this mechanism potentially originating from early eukaryotic ancestors and persisting as a functionally significant process despite variations in frequency and biological role across divergent lineages [18].
The molecular machinery facilitating trans-splicing shares remarkable conservation with canonical spliceosomal components. Evidence indicates that trans-splicing utilizes similar splicing signals and factors as alternative splicing, including snRNAs U2, U4, U5, and U6 [18]. In spliced-leader (SL) trans-splicing—a specialized form widespread in lower eukaryotes—a short noncoding exon from SL RNA is joined to the 5′-end of multiple pre-mRNAs, providing a mechanism for mRNA maturation and regulation that offers evolutionary advantages, particularly in processing polycistronic transcription units [18]. The conservation of splicing machinery across trans-splicing and cis-splicing mechanisms suggests an ancient evolutionary origin, with SL RNA potentially deriving from splicing U snRNAs in lower organisms with ancestral cis-splicing mechanisms [18].
Evolutionary Distribution and Quantitative Analysis of Trans-Splicing
The prevalence of trans-splicing exhibits remarkable variation across the eukaryotic domain, with certain lineages displaying extensive utilization while others employ it more sparingly. Comprehensive analysis of orthologous genes from completely sequenced eukaryotic genomes has revealed numerous shared features, suggesting that many RNA processing mechanisms have persisted since the last eukaryotic common ancestor (LECA) [20]. Phylogenomic reconstructions indicate that both major U2 and minor U12 spliceosomes were already present in LECA, resulting from ancient duplication events [20].
Table 1: Comparative Frequency of Trans-Splicing Across Eukaryotic Lineages
| Organism/Lineage | Trans-Splicing Frequency | Primary Type | Notable Features |
|---|---|---|---|
| Trypanosoma brucei | ~100% of genes | SL | Essential for processing polycistronic transcripts |
| Amphidinium carterae | ~100% of genes | SL | Dinoflagellate model |
| Caenorhabditis elegans | ~70% of genes | SL | Involved in growth recovery |
| Ascaris sp. | ~90% of genes | SL | Parasitic nematode |
| Adineta ricciae | ~60% of genes | SL | Rotifer species |
| Insects | ~1.58% of total genes | Inter/Intragenic | 1,627 events involving 2,199 genes |
| Vertebrates | Dramatically declined | Inter/Intragenic | Rare but physiologically significant |
The evolutionary distribution of trans-splicing demonstrates a fascinating pattern, with frequency peaking in protozoa, radiates, and protostomes before undergoing a dramatic decline in vertebrates [18]. The high percentage observed in invertebrates predominantly represents SL-type splicing, which can occur in 100% of genes in certain protists like A. carterae and K. micrum [18]. This distribution suggests that trans-splicing has experienced dynamic changes throughout eukaryotic evolution, with varying selective pressures and functional requirements shaping its utilization across lineages.
Recent genomic analyses continue to uncover new instances of trans-splicing in diverse organisms. In tunicates of the Ciona genus, which represent the closest invertebrate relatives to humans, approximately 50% of genes undergo SL trans-splicing, where a 16-nt 5′ exon of a 46-nt SL RNA joins to the trans-splice acceptor site of pre-mRNA [21]. The 5′ region upstream of the trans-splice acceptor site, termed the "outron," is discarded during this process [21]. Functional studies indicate that trans-spliced chimeric RNAs in C. elegans demonstrate higher translational efficiency than non-trans-spliced RNAs transcribed from the same gene, suggesting a potential regulatory advantage to this mechanism [21].
Molecular Mechanisms and Classification of Trans-Splicing
Fundamental Splicing Mechanisms
Trans-splicing events are broadly categorized based on the genomic origin of the participating RNA molecules. Intragenic trans-splicing occurs when pre-RNAs are transcribed from the same genomic locus, potentially producing chimeric RNAs through exon repetition, sense-antisense fusion, or exon scrambling [18]. Notable examples include the mod(mdg4) and lola genes in Drosophila, where intragenic trans-splicing generates diverse transcript isoforms [18]. Conversely, intergenic trans-splicing joins exons from separate genes, potentially located on different chromosomes, as observed in the human JAZF1-JJAZ1 chimeric RNA formed from genes on chromosomes 7 and 17 [18].
The molecular mechanism of SL trans-splicing involves precise recognition signals and splice site selection. Research in Ciona has revealed that trans-splice acceptor sites are preferentially located at the first functional acceptor site, with paired donor sites typically exhibiting weaker splicing signals [21]. Additionally, genes undergoing trans-splicing in Ciona display GU- and AU-rich 5′ transcribed regions, suggesting these sequence features may facilitate the trans-splicing mechanism [21].
Self-Splicing Group I Introns
Beyond spliceosomal trans-splicing, group I self-splicing introns represent another evolutionarily significant mechanism. These catalytic RNAs, ranging from 250-500 nucleotides, catalyze their own excision from precursor RNA without requiring spliceosomal proteins [2]. The self-splicing process occurs via two consecutive transesterification reactions initiated when an exogenous guanosine (ExoG) binds to the folded catalytic core of the ribozyme [2].
Group I introns are distributed across all domains of life, though they are notably abundant in fungi, plants, red algae, and green algae, which collectively account for approximately 90% of identified group I introns [2]. These autocatalytic elements are classified into five main groups (IA, IB, IC, ID, IE) based on conserved core domains and structural features, with further subdivision into 17 subgroups [2]. Although many group I introns self-splice efficiently in vitro, some require protein assistants with maturase functions for efficient splicing in vivo, which may be encoded by the intron itself or by host genome elements [2].
Experimental Protocols for Trans-Splicing Analysis
Transcriptome Assembly for Trans-Splicing Detection
Purpose: To comprehensively identify trans-splicing events and characterize 5′ transcribed regions (outrons) upstream of trans-splice acceptor sites.
Methodology:
- RNA-seq Library Preparation: Isolate high-quality RNA from target tissues or cell lines. For Ciona studies, 82 RNA-seq samples were utilized to ensure comprehensive coverage [21].
- Read Preprocessing: Use tools like cutadapt v1.11 to remove adapter sequences and low-quality bases from raw sequencing reads [21].
- Genome Alignment: Map preprocessed reads to the reference genome using splice-aware aligners such as STAR v2.7.9a with appropriate parameter settings for the organism [21].
- Transcript Assembly: Reconstruct transcripts using assembly tools like StringTie v1.2.3 and Scallop v0.10.4 to identify novel transcripts with extended 5′ exons or novel exons upstream of known transcripts [21].
- Gene Locus Redefinition: Combine newly identified transcripts with annotated transcripts from existing models, maintaining the total gene count while expanding transcriptional diversity [21].
Validation: Confirm trans-splicing events through:
- Specific detection of SL sequences in RNA-seq reads
- Experimental validation using RT-PCR with junction-spanning primers
- Comparison with TSS-seq data that precisely identifies transcription start sites through oligo-capping methods [21]
Identification of Trans-Splice Acceptor Sites
Purpose: To precisely map trans-splice acceptor sites and distinguish them from conventional transcription start sites.
Methodology:
- TSS-seq Data Generation: Employ oligo-capping methods to specifically replace the 5′ cap structure of mRNA with a synthetic oligoribonucleotide, enabling precise identification of mRNA 5′ ends [21].
- Read Classification: Categorize TSS-seq reads into SL trans-spliced RNAs and non-trans-spliced RNAs based on the presence or absence of 5′ SL sequences [21].
- Genome Mapping: Map classified reads to the reference genome using STAR v2.7.9a to identify trans-splice acceptor sites and transcription start sites [21].
- Open Chromatin Validation: Integrate ATAC-seq data to identify open chromatin regions as supportive evidence of true TSSs, filtering out potential artifacts [21].
- Cluster Identification: Identify TSS clusters within regions around 5′ ends of transcripts or upstream of trans-splice acceptor sites, focusing on those overlapping with open chromatin regions [21].
Bioinformatic Analysis:
- Perform local enrichment analysis of nucleotide content using 30-bp sliding windows with statistical testing via Mann-Whitney U test and FDR correction [21]
- Conduct local motif enrichment analysis using binomial tests and Fisher's exact tests to identify sequences preferentially associated with trans-splicing [21]
- Predict motif binding sites using FIMO v5.0.1 to identify potential regulatory elements [21]
Research Reagent Solutions for Trans-Splicing Studies
Table 2: Essential Research Reagents and Tools for Trans-Splicing Investigation
| Reagent/Tool | Specific Function | Application Examples | Technical Notes |
|---|---|---|---|
| STAR v2.7.9a | Splice-aware alignment of RNA-seq reads | Mapping preprocessed reads to reference genomes | Critical for identifying junction-spanning reads |
| StringTie v1.2.3 | Transcript assembly from aligned RNA-seq reads | Reconstructing transcript models including novel isoforms | Effective for identifying extended 5′ exons |
| Scallop v0.10.4 | Alternative transcript assembler | Complementary assembly to StringTie | Improves comprehensive transcript identification |
| cutadapt v1.11 | Adapter trimming and read preprocessing | Quality control of RNA-seq data | Essential for preparing clean reads for alignment |
| FIMO v5.0.1 | Motif scanning and analysis | Identifying enriched sequence motifs in trans-spliced genes | Uses statistical models to evaluate motif significance |
| TSS-seq Methodology | Precise identification of transcription start sites | Genome-wide mapping of 5′ ends of mRNAs | Employs oligo-capping to label 5′ cap structures |
| ATAC-seq Data | Identification of open chromatin regions | Validating true transcription start sites | Helps filter out technical artifacts in TSS identification |
Implications for Biocomputing Research
The natural precedent of trans-splicing across divergent eukaryotes offers valuable insights and molecular tools for biocomputing applications. The modular nature of trans-splicing, particularly the programmable specificity of group I introns, provides a blueprint for designing synthetic RNA processing systems [2]. These natural systems demonstrate how precise sequence recognition can be harnessed to create programmable molecular circuits with predictable input-output relationships.
The mechanistic understanding of trans-splicing, especially the sequence requirements for splice site recognition and the structural features of catalytic introns, informs the development of synthetic biological components. For instance, the characteristic GU- and AU-rich 5′ transcribed regions associated with trans-splicing in Ciona provide design principles for engineering efficient synthetic trans-splicing systems [21]. Similarly, the preference for trans-splice acceptor sites at the first functional acceptor site, coupled with weak paired donor sites, offers strategic guidance for positioning synthetic trans-splicing elements [21].
Biocomputing applications can leverage these natural mechanisms to create sophisticated RNA-based computing platforms. The ability of group I introns to perform precise excision and ligation reactions without protein factors makes them ideal candidates for molecular logic gates and signal processing elements. Furthermore, the extensive characterization of trans-splicing across evolutionary diverse organisms provides a rich repository of components that can be adapted, modified, and recombined to create novel biocomputing systems with enhanced capabilities and predictable behaviors.
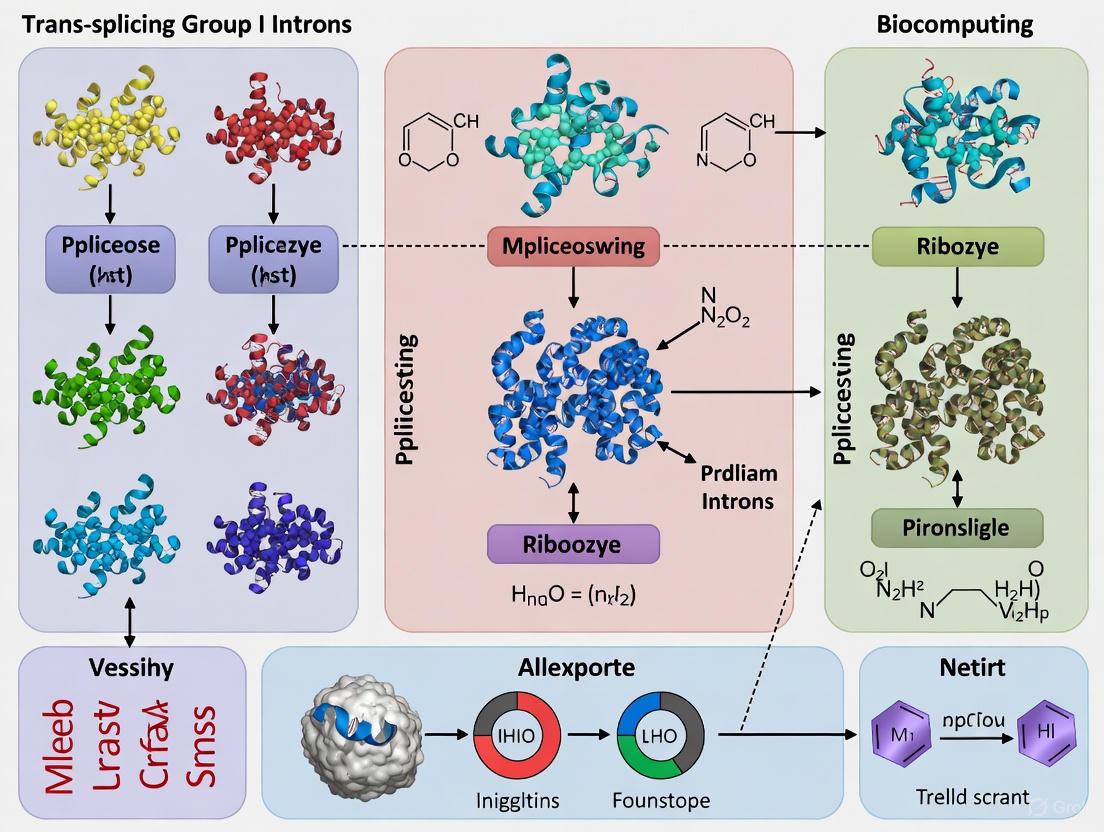
Engineering Biological Logic: Designing Trans-Splicing Ribozymes for Biocomputation and Therapy
The engineering of cis-acting ribozymes into trans-acting configurations represents a foundational principle in synthetic biology and therapeutic development. This conversion enables catalytic RNAs, which naturally act on themselves, to be reprogrammed to act on separate substrate RNAs. This principle is particularly powerful in the context of trans-splicing group I introns, which have recently gained significant attention with FDA-approved drugs entering Phase I/IIa IND trials for conditions like hepatocellular carcinoma and glioblastoma [8]. Within biocomputing research, this technology enables the construction of complex genetic circuits and programmable riboregulators, allowing for customizable, orthogonal, and predictable gene regulation [22]. This Application Note details the core engineering steps, quantitative design parameters, and experimental protocols for implementing this technology.
Core Engineering Principles and Key Design Parameters
The fundamental conversion from cis to trans involves re-engineering the ribozyme's structure to recognize an external target RNA instead of its own sequence. For the Tetrahymena thermophila group I intron, this primarily requires modifying two key recognition sequences [8].
Essential Sequence Modifications
- Internal Guide Sequence (IGS) Redesign: The native IGS, which specifies the splice site in cis, is converted into a short, trans-acting sequence. This new IGS is designed to be complementary to the target RNA, forming a duplex (the P1 helix) that positions the ribozyme's catalytic core at the correct uridylate (U) splice site on the substrate [8].
- Implementation of an Extended Guide Sequence (EGS): Trans-splicing efficiency can be dramatically increased by extending the ribozyme's 5'-terminus beyond the splice site. The EGS comprises [8]:
- A P1 extension (P1ex) of 3 nucleotides.
- An internal loop region.
- An antisense duplex of variable length (from 8 to over 40 base pairs) that enhances binding stability to the substrate.
The following table summarizes the key functional components and their design considerations.
Table 1: Core Components of a Trans-Splicing Group I Intron Ribozyme
| Component | Function | Design Consideration | Optimal Parameters / Example |
|---|---|---|---|
| Internal Guide Sequence (IGS) | Binds target RNA to define splice site via P1 helix [8]. | 6 nucleotides long; must base-pair with target sequence ending with a uridylate (U). | IGS is reverse complementary to target positions p-5 to p. |
| Splice Site Uridylate (U) | The nucleotide on the target RNA where splicing occurs [8]. | Computational prediction of accessibility is critical. | Identified via free energy calculations (e.g., using IntaRNA2) [8]. |
| Extended Guide Sequence (EGS) | Enhances splicing efficiency via increased binding stability [8]. | Includes P1ex, an internal loop, and an antisense duplex. | Antisense duplexes of 8-46 bp; EGS internal loop of 3-6 nt [8]. |
| 3'-Exon | The repair sequence or functional payload to be ligated to the target's 5'-fragment [8]. | Encodes the therapeutic gene correction or functional RNA output. | Wild-type cDNA sequence to correct a pathogenic mutation. |
Quantitative Data and Performance Metrics
The engineering process involves balancing multiple quantitative parameters to optimize ribozyme activity. The data below, derived from recent studies, provides guidance for rational design.
Table 2: Quantitative Design Parameters for Trans-Splicing Ribozymes
| Parameter | Impact on Activity | Typical Range / Value | Experimental Evidence |
|---|---|---|---|
| Splice Site Accessibility (Free Energy) | Lower (more negative) binding free energy predicts higher splicing efficiency [8]. | Computed for all candidate Us in target region. | Method from [8]; uses IntaRNA2 with --seedBP 9 parameter. |
| Antisense Duplex Length | Longer duplexes increase binding affinity but may reduce product release or cellular availability. | 8 to 46 base pairs. | Shorter duplexes (8 bp) suffice with highly accessible splice sites [8]. |
| EGS Optimization Impact | A single beneficial mutation in the EGS can dramatically enhance efficiency. | >50-fold increase possible. | Combinatorial libraries with randomized EGS identified highly active variants [8]. |
| Mutational Tolerance (Neutral Network) | The number of functional ribozyme sequences is vast, allowing for extensive engineering. | >10^39 self-reproducing sequences estimated for group I introns [23]. | Generative models (DCA) produced active variants up to 65 mutations from wild-type [23]. |
Experimental Protocol: Developing a Trans-Splicing Ribozyme
This protocol outlines the key steps for creating a trans-splicing ribozyme to repair a mutated mRNA, based on the methodology used for NF1 mRNA repair [8].
Computational Splice Site Identification
Objective: To identify the most accessible uridylate (U) splice site on the target mRNA. Procedure:
- Input Sequence: Use the target mRNA coding sequence (e.g., human NF1, NM_001042492.3).
- Free Energy Calculation: For every uridylate downstream of a chosen start position (e.g., position 4500), compute the free energy of substrate binding using IntaRNA2 [8].
- Parameter Settings:
--seedBP 9--seedQRange 1-9--seedTRange (p-5)-(p+3)(for the specific U at positionp)- Use the
turner99energy parameters from the ViennaRNA package.
- Analysis: Calculate average binding energies across multiple sequence window sizes (100-600 nt). Select the site with the most favorable (most negative) average binding energy for experimental validation.
In Vitro Validation of Splice Site and EGS
Objective: To biochemically validate the computationally predicted splice site and identify a high-efficiency EGS. Reagents:
- DNA Template: Template for a truncated version of the target RNA.
- Ribozyme Library: A pool of ribozyme constructs with a randomized EGS region and a unique barcode in the 3'-tail for deep sequencing identification [8].
- NTPs: Including [α-32P]GTP for radioactive labeling or non-radioactive alternatives.
- Transcription Buffer: e.g., from MEGAscript T7 kit.
- Reaction Buffer: 10 mM Tris, 10 mM MgCl₂.
Procedure:
- RNA Synthesis: In vitro transcribe and purify the target RNA and ribozyme library.
- Annealing: Mix 0.2-0.6 µM target RNA with a 2.5-fold molar excess of the ribozyme library in 10 mM Tris (without MgCl₂). Heat to 95°C for 2 min, then incubate at room temp for 5 min.
- Trans-splicing Reaction: Initiate the reaction by adding MgCl₂ to a final concentration of 10 mM. Incubate at 37°C for 1 hour.
- Product Analysis: Resolve the reaction products via denaturing PAGE. Identify successful trans-splicing products by their altered size.
- EGS Selection: For the EGS library screen, extract the RNA post-reaction and use high-throughput sequencing of the ribozyme's barcode to identify EGS sequences enriched in the active fraction.
Cell-Based Validation
Objective: To confirm trans-splicing activity in a relevant cellular model. Cell Line: HEK293 NF1-/- cells stably expressing a full-length mutant mNf1 cDNA [8]. Procedure:
- Transfection: Transfer the validated ribozyme (with optimal EGS) into the cell line using a standard transfection method (e.g., lipofection).
- RNA Isolation: Harvest total RNA 24-48 hours post-transfection.
- RT-PCR Analysis: Perform reverse transcription followed by PCR with primers spanning the predicted splice junction.
- Validation: Sequence the PCR product to confirm the presence of the precise, corrected mRNA sequence.
Workflow and Biocomputing Application Visualization
The following diagrams, generated with Graphviz, illustrate the core engineering workflow and a key biocomputing application.
Engineering Workflow for Trans-Splicing Ribozymes
Trans-Splicing Ribozyme in Biocomputing Logic
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Trans-Splicing Ribozyme Engineering
| Reagent / Material | Function / Application | Example / Specification |
|---|---|---|
| Tetrahymena thermophila Group I Intron Scaffold | The catalytic backbone for engineering trans-splicing ribozymes [8]. | Well-characterized sequence; used in FDA-approved drug trials [8]. |
| Computational Prediction Software | Identifies accessible splice sites on target mRNA. | IntaRNA2 with Turner '99 energy parameters [8]. |
| In Vitro Transcription Kit | Synthesizes target mRNA and ribozyme RNA for biochemical assays. | MEGAscript T7 Transcription Kit [24]. |
| Extended Guide Sequence (EGS) Library | A combinatorial pool of ribozymes with randomized EGS for efficiency optimization [8]. | Library includes a unique barcode in the 3'-tail for NGS identification. |
| Model Cell Line | Validates ribozyme function in a cellular context. | HEK293 NF1-/- cells expressing mutant mNf1 cDNA [8]. |
Synthetic biology aims to program living cells with customized functions, much like we program computers. A significant hurdle in this field has been scaling up the complexity of genetic circuits without being limited by the scarcity of reliable, non-interfering biological parts. The discovery and engineering of Split-Intron-Enabled Trans-splicing Riboregulators (SENTRs) mark a pivotal advancement in this endeavor [25] [26]. This document provides detailed application notes and protocols for utilizing SENTRs, a novel class of post-transcriptional regulators based on the programmable RNA trans-splicing activity of the group I intron ribozyme from Azoarcus [7]. SENTRs provide a versatile toolkit for constructing complex multi-input logic gates within bacterial cells, enabling sophisticated cellular computation for applications in biosensing and therapeutic intervention.
Background and Principles
The SENTR Mechanism
SENTRs are built upon the natural mechanism of group I intron ribozymes, which are catalytic RNAs that excise themselves from precursor RNA transcripts and ligate the flanking exons together. The SENTR system adapts the Azoarcus group I intron, a compact and fast-folding ribozyme, for trans-splicing applications [7]. The core innovation involves splitting the intron into two halves and fusing each half to de novo-designed External Guide Sequences (EGS) [25]. These EGSs are short RNA guides programmed to hybridize with specific target mRNAs via complementary base-pairing. This hybridization brings the split intron halves into proximity, allowing them to reassemble into a catalytically active ribozyme. The active ribozyme then performs a trans-splicing reaction, excising a portion of the target mRNA and replacing it with a new RNA sequence encoded by the SENTR's 3' exon [25] [26]. This mechanism allows for the reprogramming of gene expression at the mRNA level.
Key Features and Advantages
SENTRs exhibit several characteristics that make them ideal for building complex genetic circuits [25]:
- High Programmability: EGS sequences can be computationally designed to target virtually any mRNA.
- High Predictability: Machine learning models can predict trans-splicing activity from EGS sequences, enabling rational design.
- Low Leakage & Wide Dynamic Range: SENTRs exhibit minimal basal activity and strong activation upon target recognition.
- Orthogonality: Multiple SENTRs with distinct EGSs can function simultaneously in the same cell with minimal crosstalk, enabling parallel processing.
Application Notes: Implementing Logic Gates with SENTRs
SENTRs can be configured to perform a wide array of Boolean logic operations by sensing the presence or absence of specific input RNAs (e.g., mRNAs or synthetic small RNAs). The output is the production of a functional protein, such as a fluorescent reporter or a transcription factor, only when the logical condition is met.
Single-Layer Multi-Input Gates
A key advantage of SENTRs is their ability to process multiple inputs within a single regulatory layer. By coupling RNA trans-splicing with split intein-mediated protein trans-splicing, a single transcription factor can be controlled by multiple inputs [25]. For example, a six-input AND gate was constructed by inserting three orthogonal split introns and two orthogonal split inteins into a single gene (e.g., ecf20). Only when all six input RNAs are present do three sequential RNA splicing and two protein splicing reactions occur, producing a functional transcription activator that turns on a reporter gene [25]. This design dramatically reduces the need for multiple transcription factors required by conventional layered architectures.
The table below summarizes the performance characteristics of SENTR-based logic gates as documented in the foundational research.
Table 1: Performance Characteristics of SENTR-Based Systems
| Feature | Description / Performance Metric | Significance |
|---|---|---|
| Dynamic Range | Wide dynamic range reported [25] | Enables strong distinction between "ON" and "OFF" states. |
| Predictability | High predictability enabled by machine learning models [25] | Facilitates forward design of functional EGS guides. |
| Orthogonality | Low crosstalk with multiple orthogonal SENTR pairs [25] | Allows for independent parallel operation of multiple gates. |
| Gate Complexity | Demonstration of up to six-input AND gates [25] | Represents the most complex genetic AND circuit reported. |
| Regulatory Scope | Regulation of fluorescent proteins, transcription factors, and sgRNAs [25] | A versatile tool for controlling diverse genetic outputs. |
The Scientist's Toolkit: Essential Research Reagents
The following table lists the key biological parts and reagents required to implement SENTR-based genetic circuits.
Table 2: Research Reagent Solutions for SENTR Implementation
| Reagent / Component | Function in the System | Example / Notes |
|---|---|---|
| Azoarcus Group I Intron Fragments | The catalytic core of the SENTR system. | Split intron halves derived from the bacterial tRNAIle intron [7]. |
| External Guide Sequences (EGS) | Provides target specificity through RNA-RNA hybridization. | De novo-designed RNA sequences; design is facilitated by machine learning [25]. |
| Orthogonal SENTR Pairs | Enables independent logic channels within a single cell. | Libraries of SENTRs with low-sequence similarity EGSs to prevent crosstalk [25]. |
| Split Inteins | Enables post-translational reassembly of functional proteins. | Used in conjunction with split introns for multi-input protein-level gates (e.g., six-input AND) [25]. |
| Output Reporter Genes | Provides a measurable readout of circuit activity. | Fluorescent proteins (e.g., GFP), transcription factors (e.g., ECF20), or sgRNAs [25]. |
Experimental Protocols
Protocol 1: Designing and Testing a Novel SENTR
This protocol outlines the steps for creating a new SENTR to target a specific mRNA of interest.
Workflow Diagram: SENTR Design and Testing
Materials:
- DNA Constructs: Plasmid backbone for SENTR expression in E. coli.
- Software: Computational tools for EGS design and machine learning models for activity prediction [25].
- Host Strain: E. coli cells suitable for genetic circuit expression.
- Reagents: Primers for cloning, culture media, and reagents for RT-PCR and sequencing.
Procedure:
- Target Identification: Select the target mRNA and the specific uridine (U) residue within it where trans-splicing should occur. The only sequence requirement is a U at the splice site to base-pair with a G in the ribozyme's Internal Guide Sequence (IGS) [7].
- EGS Design: Computationally design EGS sequences that are complementary to the regions flanking the target splice site. Use available machine learning models to predict and optimize the trans-splicing efficiency of the designed EGSs [25].
- Molecular Cloning: Synthesize the SENTR construct by fusing the designed EGSs to the 5' ends of the split Azoarcus intron halves. Clone this construct into an appropriate expression plasmid downstream of a constitutive or inducible promoter.
- Host Transformation: Transform the assembled plasmid into your E. coli laboratory strain.
- Functional Validation:
- Quantitative Output: Measure the output signal (e.g., fluorescence from a spliced reporter) and compare it to negative controls to assess dynamic range and leakage.
- Splicing Verification: Confirm the correct trans-splicing event using RT-PCR to amplify the spliced product, followed by sequencing of the amplicon.
Protocol 2: Constructing a Multi-Input AND Gate
This protocol describes the assembly of a logic gate requiring multiple inputs for activation, using the six-input AND gate as a paradigm [25].
Workflow Diagram: Six-Input AND Gate Construction
Materials:
- Orthogonal SENTR Pairs: A library of at least three SENTR pairs with EGSs designed to have minimal crosstalk [25].
- Split Intein Pairs: A library of orthogonal split inteins [25].
- Output Gene: The gene to be controlled (e.g., ecf20), split and modified with intron and intein fragments.
Procedure:
- Circuit Design: Select three orthogonal SENTR pairs and two orthogonal split intein pairs from existing libraries.
- Gene Assembly: Engineer the output gene (e.g., ecf20) by inserting the three pairs of split introns and the two pairs of split inteins at strategic locations within its coding sequence. This will fragment the gene into several segments, each flanked by a splicing site.
- Plasmid Construction: Clone the entire fragmented gene construct into a single plasmid or distribute across compatible plasmids.
- Logic Evaluation: Co-express the six input RNAs that act as the triggers for the three SENTR pairs. The presence of an input RNA should trigger its corresponding RNA trans-splicing event.
- Output Measurement: Monitor the production of the functional, full-length output protein (e.g., ECF20). A high output signal should be detectable only when all six input RNAs are present, confirming the AND logic. The output can be measured by its activity (e.g., activation of a cognate promoter driving a reporter gene) or via immunoblotting.
Troubleshooting Guide
Table 3: Common Issues and Solutions in SENTR Implementation
| Problem | Potential Cause | Suggested Solution |
|---|---|---|
| High Leakage (Background) | Non-specific intron assembly or splicing. | Redesign EGS to improve specificity; use machine learning models to predict and filter out leaky designs [25]. |
| Low ON Signal (Poor Yield) | Inefficient trans-splicing or poor EGS binding. | Optimize EGS length and complementarity; ensure the Azoarcus ribozyme is in its preferred secondary structure context for trans-splicing [7]. |
| Crosstalk Between Gates | Lack of orthogonality between SENTR pairs. | Select EGS pairs with lower sequence similarity from orthogonalized libraries [25]. |
| No Splicing Detected | Incorrect splice site selection or inactive ribozyme. | Verify the target site has an accessible U residue; confirm the catalytic activity of the split intron halves in a control assay [7]. |
Therapeutic mRNA repair represents a transformative approach for treating genetic disorders at the transcript level, offering a promising alternative to conventional gene therapy. This application note focuses on the use of trans-splicing group I intron ribozymes to correct disease-causing mutations, with specific application to Neurofibromatosis Type I (NF1). This monogenic disorder results from mutations in the NF1 gene, which encodes neurofibromin, a critical regulator of the RAS signaling pathway. Loss of functional neurofibromin leads to uncontrolled cell growth and tumor formation throughout the nervous system [27] [28].
Trans-splicing ribozymes function by replacing mutated segments of mRNA with corrected sequences through precise RNA recombination events. The recent FDA approval of trans-splicing-based drugs for investigational new drug (IND) phase 1/2a trials has accelerated interest in this therapeutic modality [27]. This technology is particularly valuable for targeting large genes like NF1 (spanning over 350 kb with 60 exons) where conventional gene replacement strategies face substantial delivery challenges due to packaging limitations of viral vectors [27] [28].
Quantitative Analysis of Trans-Splicing Systems
The development of effective mRNA repair strategies requires careful selection of ribozyme systems and optimization parameters. The table below summarizes key characteristics of two prominent group I intron ribozymes used in therapeutic trans-splicing applications.
Table 1: Comparative Analysis of Group I Intron Ribozymes for Therapeutic Trans-Splicing
| Parameter | Tetrahymena thermophila Ribozyme | Azoarcus Ribozyme |
|---|---|---|
| Origin | Eukaryotic (protozoan) | Bacterial (Azoarcus BH72) |
| Size | ~400 nucleotides | ~205 nucleotides |
| Natural Context | 26S rRNA | tRNA-Ile anticodon stem-loop |
| Folding Kinetics | Slter folding in vitro | Faster folding in vitro |
| Splice Site Preference | Uracil residue required at splice site paired with 5'-terminal G of IGS | Uracil residue required at splice site paired with 5'-terminal G of IGS |
| EGS Optimization | Standard EGS design improves efficiency | Requires context resembling natural cis-splicing structure |
| In Vitro Efficiency | High under optimized conditions | Comparable to Tetrahymena when properly designed |
| Cellular Performance | Effective in mammalian cells | Reduced efficiency in E. coli cells |
The selection of appropriate splice sites on target mRNAs represents a critical design parameter. Research indicates that both Tetrahymena and Azoarcus ribozymes favor the same splice sites on a given substrate mRNA when tested in vitro, with efficiency dependent on local RNA secondary structure and accessibility [7]. The table below outlines key experimental parameters that influence trans-splicing outcomes in therapeutic contexts.
Table 2: Experimental Parameters for Optimized mRNA Repair in NF1
| Parameter | Optimized Condition | Impact on Efficiency |
|---|---|---|
| EGS Length | 100-150 nucleotides | Enhances specificity and binding affinity |
| P1 Helix Strength | Balanced stability | Prevents off-target splicing while maintaining activity |
| Magnesium Concentration | Near-physiological (≥2mM) | Essential for catalytic activity and structural stability |
| Target Site Accessibility | Computationally predicted open regions | Dramatically increases splicing yield |
| Splice Site Context | Uracil paired with ribozyme G | Absolute requirement for catalytic activity |
| Cellular Delivery | Plasmid transfection or viral vectors | AAV shows promise for in vivo applications |
Experimental Protocol: NF1 mRNA Repair Using Tetrahymena thermophila Ribozymes
Splice Site Identification and Validation
Objective: Computational identification and biochemical validation of optimal trans-splicing sites within NF1 mRNA.
Materials:
- NF1 mRNA sequence (NCBI Reference Sequence: NM_000267.3)
- RNA structure prediction software (e.g., mFold, RNAfold)
- HEK293 NF1-/- cell line
- RT-PCR reagents and gel electrophoresis equipment
- Tetrahymena thermophila group I intron ribozyme construct
Procedure:
- Computational Analysis:
- Identify potential trans-splicing acceptor sites containing uridine residues within NF1 mRNA
- Analyze RNA secondary structure around candidate sites using prediction algorithms
- Select sites with minimal secondary structure and high accessibility scores
- Design ribozyme internal guide sequences complementary to 5-12 nucleotides upstream of selected uridine splice sites
Ribozyme Construction:
- Clone Tetrahymena thermophila group I intron sequence into expression vector
- Incorporate selected guide sequences at the 5' terminus of the ribozyme
- Include Extended Guide Sequence (EGS) elements to enhance binding specificity
- Fuse corrected NF1 3' exon sequence to the 3' end of the ribozyme
Biochemical Validation:
- Transfect HEK293 NF1-/- cells with ribozyme expression constructs
- Incubate for 48-72 hours to allow trans-splicing activity
- Extract total RNA and perform RT-PCR using NF1-specific forward primer and ribozyme-specific reverse primer
- Resolve amplification products on agarose gels and sequence confirmed bands
- Quantify correction efficiency using densitometry or qPCR methods
Extended Guide Sequence (EGS) Optimization
Objective: Identification of efficiency-enhancing EGS elements through combinatorial screening.
Materials:
- EGS library with randomized sequences
- Reporter construct containing target NF1 sequence
- High-throughput sequencing capabilities
- Flow cytometry equipment for sorting (if using FACS-based selection)
Procedure:
- Library Construction:
- Generate ribozyme library with partially randomized EGS regions (20-30 nt)
- Clone library into mammalian expression vector with selectable marker
Combinatorial Selection:
- Co-transfect HEK293 cells with ribozyme library and NF1 reporter construct
- Apply selective pressure to enrich successfully repaired cells
- Recover ribozyme sequences from surviving cells after 7-14 days
- Amplify and sequence EGS regions from selected populations
- Identify consensus sequences and structural motifs associated with high efficiency
Validation:
- Test individual optimized EGS-ribozyme constructs in triplicate experiments
- Compare trans-splicing efficiency to non-optimized controls
- Evaluate specificity using RNA-Seq to detect off-target splicing events
Signaling Pathways and Workflow Visualization
The therapeutic mechanism of mRNA repair for NF1 functions through restoration of the RAS signaling pathway. The following diagram illustrates the pathological signaling in NF1 and the corrective mechanism of trans-splicing ribozymes.
The experimental workflow for developing and validating mRNA repair systems involves multiple coordinated steps from design to functional assessment, as illustrated below.
Research Reagent Solutions
The successful implementation of mRNA repair protocols requires specific reagent systems optimized for trans-splicing applications. The following table details essential research reagents and their functions in therapeutic mRNA repair workflows.
Table 3: Essential Research Reagents for mRNA Repair Studies
| Reagent Category | Specific Examples | Function & Application |
|---|---|---|
| Ribozyme Systems | Tetrahymena thermophila group I intron, Azoarcus group I intron | Catalytic RNA backbone for trans-splicing reaction; size and folding kinetics determine application suitability |
| Delivery Vectors | AAV-K55 (engineered capsid), Lentiviral vectors, Plasmid constructs | Enable cellular delivery of ribozyme constructs; engineered AAV variants provide tumor-specific targeting |
| Cell Lines | HEK293 NF1-/-, Schwann cell models, Patient-derived NF1 tumor cells | Provide biologically relevant screening systems; validate therapeutic efficacy in disease models |
| Detection Reagents | NF1-specific primers, Antibodies against neurofibromin, RAS-GTP pulldown assays | Enable quantification of trans-splicing efficiency and functional correction of RAS signaling |
| EGS Libraries | Randomized sequence libraries, Bioinformatics-optimized designs | Facilitate screening for enhanced specificity and efficiency through combinatorial approaches |
| Animal Models | Xenograft mouse models with human NF1 tumors, Genetically engineered NF1 mouse models | Provide in vivo validation of tumor suppression and therapeutic safety profiles |
The application of trans-splicing group I intron ribozymes for therapeutic mRNA repair represents a promising frontier in the treatment of monogenic disorders like Neurofibromatosis Type I. The experimental protocols outlined herein provide a framework for developing and optimizing these sophisticated molecular tools. Recent advances in ribozyme engineering, including the identification of efficiency-enhancing Extended Guide Sequences and the development of tumor-targeted delivery vectors like AAV-K55, have significantly improved the therapeutic potential of this approach [27] [29].
The integration of mRNA repair technologies with emerging biocomputing applications creates exciting opportunities for developing "smart" therapeutic systems capable of complex cellular logic operations [22]. As the field progresses, key challenges remain in optimizing delivery efficiency to extrahepatic tissues, minimizing potential immune responses, and ensuring long-term safety profiles [30]. However, with continued refinement of ribozyme design parameters and delivery systems, trans-splicing-based mRNA repair is poised to transition from experimental concept to viable clinical strategy for NF1 and other genetic disorders, ultimately fulfilling the promise of precision genetic medicine.
The increasing prevalence of fungal resistance to conventional antifungal agents necessitates the development of novel therapeutic strategies with unique mechanisms of action. This application note details a high-throughput screening (HTS) platform leveraging engineered trans-splicing group I intron ribozymes for antifungal discovery. These ribozymes, central to biocomputing research for their programmable logic capabilities [22] [25], can be designed to target and reprogram essential fungal mRNAs. Our system exploits the ribozymes' ability to perform RNA-level computation [25] by trans-splicing a reporter gene onto target fungal mRNAs, creating a direct, quantifiable readout of target viability for drug screening.
Key Principles of Trans-Splicing Ribozymes
Group I intron ribozymes are autocatalytic RNAs that naturally catalyze their own excision and the ligation of flanking exons (cis-splicing) [31]. Engineered trans-splicing variants are split into two fragments and reprogrammed to recognize a specific substrate mRNA through complementary base-pairing interactions, typically via designed External Guide Sequences (EGSs) [25]. Upon binding, the ribozyme catalyzes a trans-splicing reaction that replaces a portion of the target mRNA with a new RNA sequence, such as a reporter gene [31] [25].
The core reaction involves two transesterification steps [31]:
- The 3'-OH of an exogenous guanosine cofactor attacks the 5' splice site.
- The freed 5' exon then attacks the 3' splice site, ligating it to the ribozyme's 3' exon.
This mechanism allows for the precise, conditional repair or alteration of mRNA sequences based on the presence of a specific target, forming the basis for a highly specific biosensor [25].
Research Reagent Solutions
The following table outlines the essential components required for developing the ribozyme-based HTS assay.
Table 1: Key Research Reagents for Ribozyme-Based Screening
| Reagent Category | Specific Example/Feature | Function in the Assay |
|---|---|---|
| Engineered Ribozyme | SENTR system [25] with split intron halves from Tetrahymena thermophila [22] [31] | Core catalytic element; can be programmed via EGSs to bind and splice target fungal mRNA. |
| External Guide Sequence (EGS) | De-novo-designed RNA guide [25] | Confers target specificity by hybridizing to the fungal mRNA of interest, guiding the ribozyme to the correct splice site. |
| Reporter Exon | Fluorescent protein (e.g., GFP) or luciferase gene [25] | Spliced onto the target mRNA by the ribozyme, providing a quantifiable luminescent or fluorescent signal for HTS. |
| Guanosine Cofactor | Exogenous guanosine [31] | Essential for the first step of the trans-splicing reaction, initiating the catalytic process. |
| HTS-Compatible Detection | Fluorescence or luminescence plate reader | Enables automated, high-throughput measurement of reporter signal, indicating successful ribozyme activity and potential inhibitor presence. |
Quantitative Design Parameters
The activity and specificity of trans-splicing ribozymes are governed by several quantifiable parameters, which must be optimized for a robust HTS assay.
Table 2: Key Quantitative Parameters for Ribozyme Assay Development
| Parameter | Typical Range / Target Value | Significance & Optimization Notes |
|---|---|---|
| EGS Binding Length | 6-7 base pairs (for P9.2 helix) [31] | Shorter lengths may reduce off-target splicing; longer lengths increase specificity but may hinder ribozyme assembly. |
| Mg²⁺ Concentration | 1 mM (physiological) to 10 mM (optimized) [32] | Critical for ribozyme folding and catalysis. Cooperative activation (Hill coefficient ~1.7) observed in related ribozymes [32]. |
| Catalytic Efficiency (Vmax/Km) | Up to 3.2 × 10⁶ min⁻¹M⁻¹ (for enhanced hammerhead ribozymes) [32] | A benchmark for desired ribozyme performance. Optimized via experimental evolution and machine learning on EGS sequences [25]. |
| Orthogonality | Low sequence similarity between EGS sets [25] | Essential for multiplexed screening or targeting multiple fungal genes without cross-talk. |
| Turnover Rate | >300 nM·min⁻¹ (under substrate excess) [32] | Indicates the ribozyme's capacity for multiple turnovers, amplifying the signal in an HTS setting. |
Experimental Protocol
Ribozyme Design and Cloning
- Target Selection: Identify an essential, conserved sequence region within the target fungal mRNA (e.g., from Candida albicans or Aspergillus fumigatus).
- EGS Design: Design two EGSs complementary to the flanking regions of the target splice site. Fuse these EGSs to the split intron halves to create the SENTR ribozyme construct [25]. For initial validation, use a well-characterized ribozyme like the one from Tetrahymena thermophila [22] [31].
- Cloning: Clone the engineered ribozyme construct into an appropriate expression vector. Clone the reporter exon (e.g., luciferase) downstream of the ribozyme's 3' splice site.
HTS Workflow Implementation
The following diagram outlines the complete high-throughput screening workflow, from setup to hit identification.
- Assay Setup: Seed fungal cells in 384-well plates. Pre-treat with compounds from a small-molecule library.
- Ribozyme Delivery: Transfer the ribozyme-reporter construct into the fungal cells.
- Incubation: Incubate the plates to allow for fungal growth, ribozyme delivery, trans-splicing, and reporter gene expression.
- Signal Detection: Measure the luminescent or fluorescent signal using a plate reader. A decrease in signal relative to untreated controls indicates successful inhibition of the fungal target, as the ribozyme cannot perform splicing on dead or growth-arrested cells.
- Hit Identification: Calculate a Z'-factor > 0.5 to validate the assay's robustness [32]. Identify primary hits as compounds showing significant signal reduction at a predefined threshold (e.g., >3 standard deviations from the mean of negative controls).
Counter-Screening and Validation
- Specificity Counter-Screen: Re-test primary hits in a nearly identical assay where the ribozyme targets a non-essential fungal gene or a human mRNA ortholog. Discard compounds that show activity in this counter-screen.
- Mechanistic Validation: Confirm the mode of action of validated hits using secondary assays, such as RT-PCR to directly visualize the reduction of correctly spliced target mRNA in treated fungal cells.
Integration with Biocomputing and Advanced Applications
The underlying technology is a cornerstone of synthetic biology for its logic computation capabilities [22] [25]. A single ribozyme can be designed to process multiple inputs via orthogonal EGSs, acting as a molecular logic gate that triggers a reporter signal only when multiple essential fungal mRNAs are present [25]. This allows for the development of sophisticated screens that identify compounds disrupting specific genetic pathways or network hubs, rather than single targets, potentially leading to more resilient antifungal strategies.
The engineering of biological systems to monitor and manipulate cellular activity requires sophisticated molecular tools that can sense intracellular cues and trigger precise responses. Within the context of biocomputing research, trans-splicing group I introns have emerged as a powerful and programmable platform for creating such sensor-actuator devices. These catalytic RNA molecules can be designed to detect specific intracellular RNA sequences and, through a self-splicing mechanism, link this detection to the production of orthogonal protein outputs. This application note details the principles, quantitative performance metrics, and standardized protocols for implementing group I intron-based RNA sensors, providing researchers with a practical framework for integrating these devices into synthetic gene networks and therapeutic applications.
Key Principles and Design Rules
Group I introns are catalytic RNAs (ribozymes) that naturally undergo cis-splicing, excising themselves from primary transcripts and ligating the flanking exons [3]. Engineering these elements into trans-splicing devices involves splitting the intron and exons such that the ribozyme can assemble on and reprogram a separate target RNA substrate [31] [25].
- Sensor Mechanism (RNA Detection): The sensor function is achieved by programming the ribozyme's Internal Guide Sequence (IGS) or attaching de-novo-designed External Guide Sequences (EGSs). These sequences base-pair with a specific target RNA molecule within the cell [25]. This interaction brings the splice sites into proximity for the catalytic core.
- Actuator Mechanism (Protein Output): The actuator function is encoded within the ribozyme's 3' exon, which can contain sequences for a desired output protein (e.g., a fluorescent reporter, transcription factor, or therapeutic protein) [31]. Upon detection and binding to the target RNA, the ribozyme catalyzes a trans-splicing reaction, precisely replacing a portion of the target RNA with its own 3' exon and thereby enabling translation of the encoded output protein [7].
The core design can be adapted for different logical operations. For instance, by designing EGSs to hybridize with intracellular mRNAs or synthetic small RNAs, systems have been created to implement Boolean logic functions like AND, NAND, and NOR gates [25].
Performance Data and Device Characterization
The following tables summarize key performance characteristics of different group I intron ribozymes and related technologies, providing a basis for selection and engineering.
Table 1: Comparative Analysis of Group I Intron Ribozymes for Trans-Splicing
| Ribozyme Source | Size (nt) | Subgroup | Optimal 5' Design | Relative Splicing Efficiency (in vitro) | Key Characteristic |
|---|---|---|---|---|---|
| Tetrahymena thermophila [31] [7] | ~400 | IC1 | Extended Guide Sequence (EGS) | High (Benchmark) | Robust activity; well-characterized. |
| Azoarcus BH72 [7] | ~205 | IC3 | tRNA anticodon stem-loop mimic | Similar to Tetrahymena (in vitro) | Fast folding; compact size. |
| Scytalidium dimidiatum [33] | - | - | - | - | Low innate immune activation; suitable for vaccines. |
Table 2: Performance Metrics of RNA-Sensing Platforms
| Technology Platform | Core Mechanism | Key Output | Reported Fold Induction | Therapeutic Context Demonstrated |
|---|---|---|---|---|
| Trans-splicing Group I Intron [31] [25] | Target RNA-guided splicing | Protein translation | - | - |
| ADAR-Based (CellREADR) [34] | A-to-I RNA editing | Protein translation | - | Cell type-specific monitoring and manipulation |
| Intrabody-Based Protein Sensor [35] | Protein-induced TEV protease cleavage | Transcriptional activation | Up to 100-fold | HCV, Huntington's disease, HIV |
Experimental Protocols
Protocol: Designing a Trans-Splicing Group I Intron Sensor-Actuator
This protocol outlines the steps for creating a ribozyme that detects a specific mRNA and responds by producing a fluorescent protein.
1. Sensor Design and Vector Construction
- Identify Target Sequence: Select a unique, accessible region within the target mRNA. Tools like the CellREADR portal (www.cellreadr.com) can assist in the design process [34].
- Design the IGS/EGS: Generate the reverse complement of the target sequence to create the guide RNA. The P1 helix must contain a Uracil at the target splice site paired with a Guanine in the ribozyme's IGS [7].
- Clone into Expression Vector: Assemble the following components into a suitable mammalian expression vector:
- 5' Homology Domain (E1): Fused to the 5' end of the ribozyme, complementary to the target RNA.
- Group I Intron Core: The catalytic ribozyme sequence (e.g., from Tetrahymena).
- 3' Exon (E2): Encodes the output protein (e.g., EYFP), followed by a poly-A tail.
2. In Vitro Validation of Splicing
- In Vitro Transcription: Linearize the plasmid and use a T7 High Yield RNA Synthesis Kit to produce the precursor RNA.
- Circularization Reaction: Purify the RNA and incubate in splicing buffer (e.g., 15 mM MgCl₂, 25 mM HEPES pH 7.5, 2 mM GTP) at 37°C for 15-60 minutes [33].
- Validation: Analyze the reaction products using agarose gel electrophoresis. A successful splicing reaction will produce a circular RNA product of the expected size.
3. Cell Culture Transfection and Validation
- Deliver Construct: Co-transfect the ribozyme construct and a plasmid expressing the target mRNA into HEK293T cells.
- Measure Output: After 24-48 hours, analyze cells for fluorescence output via flow cytometry or microscopy.
- Specificity Controls: Include cells transfected with the ribozyme alone and a non-target mRNA to confirm response is specific to the intended target.
Protocol: Implementing Multi-Input Logic Gates
This advanced protocol describes configuring split introns for a two-input AND gate [25].
1. Circuit Design
- Split the Intron: Divide the group I intron into two halves.
- Fuse with EGS: Attach a unique EGS to each intron half. Each EGS is designed to bind a distinct input RNA (Input A and Input B).
- Arrange Exons: Configure the system so that a functional output protein (e.g., a transcription factor) is only produced when both input RNAs are present and the split intron trans-splices correctly.
2. Assembly and Transformation
- Clone the two split-intron fragments and the output reporter construct (e.g., a fluorescent protein under the control of the transcription factor activated by the spliced product) into separate expression vectors or a single multi-cistronic vector.
- Co-transform the constructs into the target bacterial or mammalian cells.
3. Logic Gate Validation
- Test all input combinations: Measure the output signal in the presence of (1) No input, (2) Input A only, (3) Input B only, and (4) Both Input A and Input B.
- A high output only in condition (4) confirms AND gate operation. Use flow cytometry for quantitative single-cell analysis.
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions
| Reagent / Resource | Function / Description | Example Source / Identifier |
|---|---|---|
| Group I Intron Vectors | Backbone plasmids for engineering trans-splicing ribozymes. | Addgene (e.g., #192063, #192064) [34] |
| T7 High Yield RNA Synthesis Kit | For in vitro transcription of ribozyme precursor RNA. | New England Biolabs (NEB) [33] |
| HEK293T Cells | A robust mammalian cell line for transient transfection and device testing. | ATCC (CRL-3216) [34] |
| In-Fusion Snap Assembly Master Mix | For seamless, directional cloning of DNA fragments. | Takara Bio [34] |
| CellREADR Design Portal | A web-based tool for designing RNA sensors based on ADAR, a related technology. | www.cellreadr.com [34] |
Visual Synthesis
Core Mechanism of a Trans-Splicing Sensor-Actuator
Experimental Workflow for Device Validation
Optimizing Ribozyme Performance: A Guide to Enhancing Efficiency and Specificity
This application note details computational and experimental methodologies for optimizing trans-splicing group I intron efficiency, a critical technology platform for biocomputing research and therapeutic development. We present integrated protocols for predicting splice site accessibility through binding free energy calculations (ΔGbind) and empirically validating trans-splicing efficiency using fluorescence-based reporter systems. The framework enables researchers to identify optimal target sites on mRNA substrates, design high-efficiency trans-splicing ribozymes, and quantify splicing efficiency in high-throughput screening formats. These approaches address the fundamental challenge of low trans-splicing efficiency that has limited biomedical applications of group I intron ribozymes.
Group I introns are catalytic RNAs (ribozymes) that excise themselves from primary transcripts through a two-step transesterification mechanism [36] [2]. These autocatalytic introns have been engineered into trans-splicing ribozymes capable of replacing the 3'-terminal portion of an external mRNA with their own 3'-exon [37]. This molecular reprogramming capability creates powerful opportunities for biocomputing applications including logic gates, sensors, and molecular computation devices.
The trans-splicing reaction initiates when the ribozyme's Internal Guide Sequence (IGS) hybridizes to a complementary target site on a substrate mRNA, forming a helix equivalent to the P1 duplex in wild-type introns [36] [37]. The ribozyme then catalyzes cleavage of the substrate at the target site and transfer of the ribozyme 3'-exon to the remaining 5' portion of the substrate [37]. This precise molecular editing function enables the programming of RNA-based computing elements with diverse functionalities.
Despite this potential, technical challenges have limited implementations. A primary obstacle is low trans-splicing efficiency - typically 10% or less in cellular environments [37]. Efficiency varies dramatically with the location of the splice site within the mRNA substrate, largely due to secondary structures that can render potential target sites inaccessible to the ribozyme [37]. This application note establishes standardized methodologies to overcome these limitations through computational prediction and empirical validation.
Computational Prediction of Splice Site Accessibility
Theoretical Framework
The binding free energy (ΔGbind) represents the overall energy change when a ribozyme binds to a specific splice site on an mRNA substrate. This parameter directly influences trans-splicing efficiency because accessible sites with favorable (negative) ΔGbind values permit more stable ribozyme-substrate complexes [37]. The binding process can be modeled as three molecular events, each with associated energy changes:
- ΔGunfold-target: Free energy required to unfold secondary structures around the target site on the mRNA substrate
- ΔGrelease-IGS: Energy cost to release the IGS from any base pairs with the ribozyme's 3'-exon
- ΔGhybrid: Free energy change when the released IGS hybridizes with the target site to form the P1 duplex
The overall binding free energy is computed as: ΔGbind = ΔGunfold-target + ΔGrelease-IGS + ΔGhybrid [37]. Sites with strongly negative ΔGbind values typically yield higher trans-splicing efficiencies.
Calculation Protocol
Materials and Software Requirements
- RNA folding algorithms: RNAstructure, UNAFold, or ViennaRNA Package
- Computational resources: Standard desktop computer sufficient for most targets
- Input sequences: Target mRNA sequence in FASTA format; ribozyme IGS and 3'-exon sequences
Step-by-Step Procedure
Input Sequence Preparation: Obtain complete target mRNA sequence. Define all potential splice sites (typically GU dinucleotides in appropriate context for group I introns).
Secondary Structure Prediction: For each candidate splice site, compute the native secondary structure of the substrate region spanning approximately 150 nucleotides centered on the target site using partition function calculations.
Component Energy Calculations:
- Calculate ΔGunfold-target using the difference in free energy between folded and unfolded states of the target region
- Compute ΔGrelease-IGS by predicting secondary structures of the ribozyme with and without IGS-3'-exon pairing
- Determine ΔGhybrid from the predicted stability of the P1 duplex between IGS and target site
ΔGbind Computation: Sum the three component energy values for each candidate splice site.
Site Ranking: Sort potential splice sites by ΔGbind (most negative to least negative) to identify the most promising targets for experimental testing.
Table 1: Example ΔGbind Calculations for Candidate Splice Sites on CAT mRNA
| Site Position | ΔGunfold-target (kcal/mol) | ΔGrelease-IGS (kcal/mol) | ΔGhybrid (kcal/mol) | ΔGbind (kcal/mol) | Predicted Efficiency |
|---|---|---|---|---|---|
| 124 | +4.2 | +2.1 | -12.8 | -6.5 | High |
| 256 | +6.8 | +2.1 | -10.2 | -1.3 | Medium |
| 387 | +8.5 | +2.1 | -9.1 | +1.5 | Low |
Validation and Performance
This computational approach demonstrates strong correlation with experimentally measured trans-splicing efficiency. In validation studies using chloramphenicol acetyl transferase (CAT) mRNA, computed ΔGbind values showed better correlation with actual trans-splicing efficiency than experimental trans-tagging assays [37]. The method successfully identifies sites with favorable energy profiles while filtering structurally inaccessible targets.
Experimental Measurement of Trans-Splicing Efficiency
Fluorescence-Based Reporter System
Principle
Engineered group I introns from pathogenic fungi such as Fusarium oxysporum can be adapted as trans-splicing ribozymes with their activity monitored in real-time using fluorescence resonance energy transfer (FRET) pairs [36]. Successful trans-splicing brings fluorophores into proximity, generating measurable FRET signals that correlate with splicing efficiency.
Reagent Preparation
- Ribozyme Construction: Convert native group I intron to trans-splicing format by removing 5' portion to expose IGS at 5' terminus [36]
- Reporter Substrate: Design mRNA substrate containing complementary target site followed by acceptor fluorophore sequence
- FRET Pair Selection: Cy3 (donor) and Cy5 (acceptor) provide optimal spectral properties for most applications
Experimental Workflow
- In Vitro Transcription: Generate ribozyme and substrate RNAs using T7 RNA polymerase with purified PCR templates
- RNA Purification: Denaturing PAGE purification ensures structural homogeneity
- Splicing Reactions: Combine ribozyme and substrate (1:10 ratio) in splicing buffer (50mM Tris-HCl, pH 7.5, 100mM NH4Cl, 5mM MgCl2)
- Fluorescence Monitoring: Measure FRET emission (665nm) with excitation at 550nm over 60 minutes at 37°C
- Data Analysis: Calculate initial rates from linear phase of fluorescence increase; normalize to control reactions
Figure 1: Experimental workflow for trans-splicing efficiency measurement using fluorescence-based reporter system
Protocol Modifications for High-Throughput Screening
For biocomputing applications requiring testing of multiple ribozyme-substrate combinations:
- Microplate Format: Scale down reactions to 20μL in 384-well plates
- Automated Liquid Handling: Implement for reproducible reagent dispensing
- Positive Controls: Include known efficient ribozyme-substrate pairs in each plate
- Z'-Factor Calculation: Determine assay quality using (1 - (3σc+ + 3σc-)/|μc+ - μc-|) where c+ and c- are positive and negative controls
Table 2: Research Reagent Solutions for Trans-Splicing Experiments
| Reagent | Function | Storage | Quality Control |
|---|---|---|---|
| T7 RNA Polymerase | In vitro transcription of ribozyme and substrate | -80°C in 50% glycerol | RNase-free; >90% purity |
| Splicing Buffer (10X) | Provides optimal ionic conditions for ribozyme activity | -20°C | Filter-sterilized; Mg²⁺ concentration verified |
| Guanosine Cofactor | Initiates transesterification reaction | -20°C as 100mM stock | HPLC purified; dissolved in DMSO |
| Fluorescently-labeled Oligonucleotides | FRET-based detection of splicing | -80°C, protected from light | PAGE purification; concentration verified |
Integration of Computational and Experimental Approaches
Validation of Predictive Models
To establish correlation between computational predictions and experimental results:
- Select Diverse Sites: Choose 10-15 splice sites spanning a range of predicted ΔGbind values
- Experimental Testing: Measure trans-splicing efficiency for each site using fluorescence assay
- Correlation Analysis: Plot experimental efficiency against computed ΔGbind
- Model Refinement: Adjust energy parameters if systematic deviations are observed
This validation approach typically reveals correlation coefficients (R²) of 0.7-0.9 between predicted ΔGbind and measured efficiency [37], confirming the utility of computational predictions for screening potential target sites.
Application to Biocomputing Circuit Design
For complex biocomputing systems requiring multiple orthogonal ribozyme components:
- Target Site Identification: Compute ΔGbind for all potential splice sites across target transcripts
- Specificity Assessment: Evaluate cross-reactivity potential through IGS complementarity analysis
- Efficiency Ranking: Select sites with favorable ΔGbind and minimal off-target interactions
- Modular Construction: Design ribozyme components with standardized flanking sequences for assembly
This integrated approach enables rational design of RNA-based computing elements with predictable performance characteristics, moving beyond trial-and-error optimization.
Troubleshooting Guide
Table 3: Common Experimental Issues and Solutions
| Problem | Potential Causes | Solutions |
|---|---|---|
| Low FRET signal | Poor ribozyme folding | Optimize Mg²⁺ concentration; implement thermal renaturation |
| High background fluorescence | Non-specific cleavage | Increase stringency with higher temperature or formamide |
| Variable replicate results | RNA degradation | Ensure RNase-free conditions; use fresh RNA preparations |
| Poor correlation with predictions | Incorrect structural models | Include co-transcriptional folding in predictions |
| Inefficient splicing despite favorable ΔGbind | Kinetic traps in folding | Add peripheral sequences to stabilize active conformation |
The integration of computational prediction through binding free energy calculations with empirical validation using fluorescence-based reporters provides a robust framework for optimizing trans-splicing group I intron efficiency. These methodologies enable researchers to move beyond random screening approaches to rational design of ribozyme components for biocomputing applications. As the field advances, these protocols will support the development of increasingly sophisticated RNA-based computing systems with enhanced reliability and performance characteristics.
External Guide Sequence (EGS) technology represents a powerful RNA-based tool for the precise manipulation of gene expression, a capability of paramount importance to the field of biocomputing. This technology harnesses the endogenous ribonuclease P (RNase P) complex, a ubiquitous ribozyme found in all living organisms, to direct the cleavage of specific target messenger RNA (mRNA) molecules [38]. In biocomputing, which seeks to use biological components for computational operations, the ability to logically control cellular processes is a fundamental requirement. EGS technology provides a programmable "switch" for gene circuits by enabling the targeted degradation of mRNA transcripts encoding key regulatory proteins. When an EGS RNA is designed to be complementary to a specific target mRNA, it binds and forms a complex that mimics the natural substrate of RNase P, thereby eliciting cleavage and inactivation of the target transcript [38]. This precise, protein-independent mechanism allows researchers to construct sophisticated genetic networks within cells, forming the basis for cellular sensors, biological processors, and engineered living systems [39].
The relevance of EGS technology is further amplified when integrated with other RNA regulatory systems, such as trans-splicing group I intron ribozymes. These ribozymes are catalytic RNAs that can be engineered to perform trans-splicing reactions, replacing the 3' or 5' portion of a substrate RNA with an exon carried by the ribozyme itself [7] [31]. The efficiency and specificity of both EGS and trans-splicing systems can be profoundly enhanced by the strategic design of their Extended Guide Sequences (EGS), which are elongated regions facilitating stronger and more specific binding to the target RNA [7]. The synergy between these systems opens avenues for complex biocomputing operations, including signal integration, state memory, and the implementation of Boolean logic within living cells, thereby pushing the frontiers of synthetic biology and programmable cellular therapeutics.
Fundamental Principles of EGS Design
The core principle of EGS technology hinges on the natural function of RNase P, which is primarily responsible for the 5'-end maturation of transfer RNAs (tRNAs). The enzyme recognizes the tertiary structure of its substrate; any RNA molecule that can form a short duplex resembling the acceptor stem and T-stem-loop of a pre-tRNA can become a substrate for cleavage [38]. An EGS is an antisense oligoribonucleotide designed to bind a target mRNA and, through this binding, induce the formation of an RNase P-recognition structure.
A standard, highly effective EGS design for use in bacteria comprises two critical segments [38]:
- An Antisense Sequence: A segment of 13–16 nucleotides that is complementary to a chosen accessible region on the target mRNA. This base-pairing provides the specificity that directs the EGS to its intended target.
- The RCCA Sequence: The addition of the nucleotides RCCA (where R is a purine) at the 3' end of the EGS. This sequence is crucial as it facilitates the proper interaction with RNase P, making the mRNA-EGS complex a competent substrate for cleavage.
For trans-splicing group I introns, the design logic of the EGS is adapted to the specific ribozyme's architecture. The ribozyme recognizes its target site on a substrate RNA primarily through base-pairing interactions. The most common design utilizes the P1 helix, where the ribozyme's 5' terminus (the internal guide sequence, or IGS) pairs with the sequence upstream of the 5'-splice site on the substrate [7] [31]. The efficiency of this initial docking step is a key determinant of overall splicing efficiency. An Extended Guide Sequence (EGS) in this context is an elongation of the ribozyme's 5' terminus, creating a stronger and more extensive hybridization region with the substrate mRNA [7]. It is important to note that different group I introns, such as those from Tetrahymena thermophila and Azoarcus, have distinct structural preferences for their optimal EGS designs, often reflecting their natural cis-splicing context [7].
Table 1: Core Components of an Effective EGS for RNase P Recruitment
| Component | Optimal Length/Nature | Primary Function |
|---|---|---|
| Antisense Binding Arm | 13-16 nucleotides [38] | Provides specificity by binding accessible region of target mRNA. |
| 3' RCCA Sequence | 4 nucleotides (RCCA) [38] | Promotes recognition and cleavage by the RNase P holoenzyme. |
| Overall Construct | RNA, or nuclease-resistant analogs (e.g., PPMO, LNA/DNA) [38] | Acts as the structural guide for RNase P. |
Application Notes: EGS in Practice
Target Site Selection and Validation
The first and most critical step in deploying EGS technology is identifying accessible binding sites on the target mRNA. The secondary and tertiary structure of the mRNA can hide potential target sequences, making them inaccessible to EGS binding. Several empirical methods are used for this purpose, and their results can be refined using computational predictions [38].
- RNase H Mapping: This is a commonly used method where a library of DNA oligonucleotides is incubated with the target mRNA. DNA-RNA hybrids formed at accessible sites are cleaved by RNase H. The cleavage products are analyzed to map the "open" regions suitable for EGS design [38].
- In Vivo Mapping with Dimethyl Sulfate (DMS): DMS modifies accessible adenosine and cytosine residues in RNA. Cells are treated with DMS, the RNA is extracted, and the modification sites are identified by reverse transcription, revealing nucleotides that are single-stranded and thus accessible in the cellular environment [38] [7].
- EGS Library Screening: A more direct approach involves testing a pool of randomized EGSs for their ability to cleave the target mRNA in vitro or in vivo. The sequences of the most active EGSs are then determined, directly revealing effective target sites [38].
Once a target region is identified, the EGS is designed to be fully complementary to it. The sequence should be checked for specificity to minimize off-target effects within the transcriptome. Software like mfold can be used to model the EGS-mRNA hybrid to ensure it does not form internal structures that would inhibit RNase P recognition [38].
EGS-Dependent Ribozyme Engineering for Biocomputation
In biocomputing, EGS-enhanced trans-splicing ribozymes can be engineered as components of logic gates. For example, the expression of the ribozyme itself can be placed under the control of one promoter (Input A), while the expression of its specific EGS can be controlled by a second promoter (Input B). The corrective splicing event, and thus the output (a reporter or therapeutic protein), only occurs when both components are present, effectively creating an AND gate.
Table 2: Comparison of Group I Intron Ribozymes for Trans-Splicing Applications
| Ribozyme Source | Size (nt) | Optimal EGS Context | Splicing Efficiency | Key Characteristic |
|---|---|---|---|---|
| Tetrahymena thermophila | ~400 [7] | Elongated 5' terminus [7] | High in vitro and in cells [31] | Robust, well-characterized, versatile. |
| Azoarcus | ~205 [7] | Resembles natural tRNA anticodon stem-loop [7] | High in vitro, lower in cells [7] | Small size, fast folding kinetics. |
| Pneumocystis carinii | - | Utilizes P10 and P9.0 helices [31] | Effective in vitro [31] | Uses alternative 3'-splice site recognition. |
The following diagram illustrates the logical workflow for designing and implementing an EGS-based genetic circuit for biocomputing.
Experimental Protocols
Protocol 1: Evaluating EGS Efficacy in an E. coli Model System
This protocol outlines the steps for testing the activity of a candidate EGS designed to inhibit a specific gene in E. coli [38].
Principle: A recombinant plasmid expressing the EGS is introduced into E. coli cells. Successful RNase P-mediated cleavage of the target mRNA will lead to a reduction in the corresponding protein levels, which can be measured via a phenotypic assay (e.g., loss of antibiotic resistance) or directly by western blot.
Materials:
- Plasmid Construct: A plasmid containing a T7 promoter, the EGS sequence, a core hammerhead ribozyme sequence, and a T7 terminator (T7p–EGS–HH–T7t). The hammerhead ribozyme ensures self-cleavage and release of the mature EGS upon transcription [38].
- Control Plasmid: A plasmid expressing a scrambled, non-functional EGS sequence.
- Bacterial Strain: An appropriate E. coli strain, potentially including an RNase P temperature-sensitive mutant for validation of mechanism.
- Antibiotics: For selection of plasmids and for phenotypic assays (e.g., ampicillin if targeting blaTEM).
- Equipment: Standard molecular biology equipment for bacterial culture, transformation, and protein or phenotypic analysis.
Procedure:
- Clone EGS Expression Cassette: Clone the DNA sequence encoding your EGS and the scrambled control into the plasmid vector.
- Transform E. coli: Transform the recombinant plasmids into your E. coli host strain. Include a negative control (empty vector).
- Induce EGS Expression: Grow transformed cultures to mid-log phase and induce the expression of the EGS from the T7 promoter (e.g., with IPTG).
- Measure Target Knockdown:
- Phenotypic Assay: If targeting an antibiotic resistance gene (e.g., blaTEM for ampicillin resistance, cat for chloramphenicol resistance), plate induced cultures on media containing the corresponding antibiotic. A significant reduction in colony-forming units (CFUs) compared to the control indicates successful EGS activity [38].
- Biochemical Assay: Perform western blot analysis or enzymatic assays to directly quantify the reduction in the target protein level.
- Validate RNase P Dependence (Optional): Repeat the experiment with the RNase P temperature-sensitive mutant strain at the restrictive temperature. Loss of EGS activity at the restrictive temperature confirms that the observed effect is mediated by RNase P [38].
Protocol 2: Testing Trans-Splicing Ribozyme Activity with an EGS In Vitro
This protocol describes a method to quantify the efficiency of a trans-splicing group I intron ribozyme, whose target binding is facilitated by an EGS, in a cell-free system [7].
Principle: The ribozyme and its target substrate RNA are synthesized and purified. When incubated together under permissive conditions, the ribozyme will catalyze a splicing reaction that joins its 3' exon to the 5' portion of the substrate. The products are analyzed by gel electrophoresis to determine splicing efficiency.
Materials:
- Purified Ribozyme RNA: Engineered trans-splicing ribozyme (e.g., from Tetrahymena or Azoarcus) with an optimized EGS at its 5' terminus.
- Purified Substrate RNA: The target mRNA fragment containing the binding site for the EGS.
- Reaction Buffer: Typically 50 mM HEPES (pH 7.5), 5-50 mM MgCl₂, and 150 mM NH₄Cl. Mg²⁺ is a critical cofactor for ribozyme folding and catalysis.
- Nucleotide Triphosphates: For ribozymes requiring exogenous Guanosine (G), include GTP.
- Equipment: Thermocycler or water bath, polyacrylamide gel electrophoresis (PAGE) apparatus, and phosphorimager or gel documentation system.
Procedure:
- Prepare Reaction Mixture: In a microcentrifuge tube, mix:
- 50-100 nM of substrate RNA.
- 200-500 nM of ribozyme RNA (to drive the reaction).
- 1X reaction buffer.
- 1 mM GTP (if required).
- RNase-free water to volume.
- Incubate: Place the reaction mixture in a thermocycler or water bath. A common condition is 37°C for 30-60 minutes. Include a negative control without the ribozyme.
- Stop the Reaction: Add an equal volume of stop solution (e.g., 95% formamide, 10 mM EDTA, with tracking dyes).
- Analyze Products: Denature the samples at 95°C for 3 minutes and immediately load them onto a denaturing polyacrylamide gel. Run the gel at sufficient voltage to resolve the substrate from the ligated product (5' substrate fragment joined to the ribozyme's 3' exon).
- Visualize and Quantify: Stain the gel with SYBR Gold or a similar fluorescent RNA stain. Image the gel and quantify the band intensities. Splicing efficiency is calculated as the percentage of the product band intensity relative to the total (substrate + product) intensity.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Research Reagent Solutions for EGS and Trans-Splicing Experiments
| Reagent/Material | Function | Example/Note |
|---|---|---|
| EGS Expression Plasmid | For in vivo expression of EGS RNA; contains promoter, EGS, and terminator. | T7p–EGS–HH–T7t construct for self-cleaving EGS release in E. coli [38]. |
| Chemically Synthesized EGS | For RNP delivery or in vitro studies; avoids transcriptional bias. | Synthetic gRNAs show fewer sequence-based efficiency issues than transcribed ones [40]. |
| RNase P Enzyme | The catalytic ribonucleoprotein that cleaves the target mRNA-EGS complex. | Can be purified from bacterial (e.g., M1 RNA + C5 protein) or eukaryotic sources [38]. |
| Nuclease-Resistant EGS Analogs | Increases in vivo stability and efficacy. | Phosphorodiamidate Morpholino Oligomers (PPMOs) conjugated to cell-penetrating peptides [38]. |
| Group I Intron Ribozyme | The catalytic RNA core for trans-splicing operations. | Tetrahymena thermophila ribozyme is robust; Azoarcus is small and fast-folding [7] [31]. |
| In Vitro Transcription Kit | For synthesizing ribozyme and substrate RNAs for in vitro assays. | T7, SP6, or T3 RNA polymerase-based systems can be used. |
| Structure Prediction Software | To model RNA secondary structure and predict accessible target sites. | Software like mfold or RNAfold is used to refine EGS design [38]. |
In synthetic biology and biocomputing, the goal of engineering multi-component systems that function predictably relies on the principle of orthogonality—where biological parts operate independently without unwanted interference, or "crosstalk." For ribozymes, particularly the group I intron class used in trans-splicing applications, achieving orthogonality is paramount for developing complex genetic circuits, biosensors, and therapeutic tools. Trans-splicing group I introns are catalytic RNAs that can be engineered to recognize and re-write specific substrate mRNAs, making them powerful for programming cellular behavior [31] [17]. However, in systems where multiple ribozymes are deployed, a key challenge is that non-cognate ribozyme-substrate interactions can occur, leading to erroneous outputs and system failure. This application note details quantitative strategies and protocols to minimize such crosstalk, enabling the construction of robust, multi-channel ribozyme networks for advanced biocomputing research.
Quantitative Characterization of Ribozyme Crosstalk
Crosstalk arises when a ribozyme intended to target a specific substrate sequence exhibits activity towards off-target substrates. Quantifying this crosstalk is the first step in engineering orthogonality.
Table 1: Key Parameters for Quantifying Ribozyme Crosstalk
| Parameter | Description | Measurement Method | Impact on Orthogonality |
|---|---|---|---|
| Splicing Efficiency (A) | The maximum fraction of ribozyme molecules that undergo successful splicing. Often <1 due to misfolding [41]. | Fluorescence-based splicing assay after long reaction time [42]. | Lower amplitude necessitates higher ribozyme expression, potentially increasing crosstalk. |
| Rate Constant (k_cat) | The catalytic rate of the ribozyme's splicing reaction. | Measure reaction progress over time under single-turnover conditions [41]. | A high kcat/kback ratio is required for efficient isolation and clear signal over background. |
| Background Rate (k_back) | The uncatalyzed rate of the reaction in the absence of the functional ribozyme structure. | Measure reaction progress with a non-functional, scrambled ribozyme sequence [41]. | A high kcat/kback ratio is required for efficient isolation and clear signal over background. |
| Dynamic Range | The fold difference in output (e.g., fluorescence) between the fully "on" (with input) and "off" (without input) states. | Fluorescence-activated cell sorting (FACS) or plate reader measurement [42]. | High dynamic range is indicative of low crosstalk; engineered systems have achieved ~93-fold ranges [42]. |
The principles of crosstalk minimization are shared across synthetic biology. In quorum-sensing systems, for instance, quantitative models have shown that simply manipulating the expression levels of receiver proteins can influence crosstalk by 10 to 100-fold [43]. Similarly, for ribozymes, controlling concentration and reaction conditions is critical. Furthermore, thermodynamic models that guide the design of RNA-RNA interaction energies between the ribozyme's Internal Guide Sequence (IGS) and the substrate can predict and minimize off-target binding [42].
Experimental Protocols for Engineering Orthogonal Ribozyme Systems
Protocol 1: High-Throughput Identification of Orthogonal Split Sites
This protocol uses transposon mutagenesis to systematically identify ribozyme split sites that minimize un-templated assembly (a primary source of crosstalk) while maintaining high templated activity [42].
- Library Generation via Transposon Mutagenesis:
- Use a MuA transposase to insert a synthetic transposon into every possible position within the gene encoding the Tetrahymena thermophila group I intron ribozyme.
- Use PCR to replace the transposon at each site with two complementary RNA guide sequences. This creates a library of "split ribozymes" where the two fragments are brought together by the guide interaction.
- Screening with an Inducible Inhibitor:
- Clone the split ribozyme library into a plasmid where splicing restores a reporter gene (e.g., sfGFP).
- Co-transform the library into E. coli along with a second plasmid expressing an RNA inhibitor. This inhibitor is a reverse complement to one guide strand and uses a toehold displacement strategy to competitively inhibit split ribozyme assembly in the "off" state.
- Use Fluorescence-Activated Cell Sorting (FACS) to separate cells exhibiting high fluorescence (high "on" state, without inhibitor) from those with low fluorescence (low "off" state, with inhibitor).
- Next-Generation Sequencing (NGS) and Analysis:
- Sequence the sorted libraries using NGS to identify the genomic positions of split sites that yield the highest enrichment ratios (on/off signal).
- Key Insight: Functional split sites are often located in surface-accessible, low-sequence-conservation regions of the ribozyme, such as the P9 domain, while the conserved catalytic core is less tolerant of splitting [42].
Protocol 2: Minimizing Crosstalk via Directed Evolution of Substrate Recognition
This protocol describes an in vitro selection strategy to evolve ribozyme variants with enhanced specificity for their target substrate.
- Design of Randomized Substrate Recognition Pool:
- Clone a pool of trans-splicing ribozymes where the Internal Guide Sequence (IGS), which forms the P1 helix with the substrate, is partially randomized.
- This pool is designed to target a specific U nucleotide (the splice site) within a model substrate mRNA (e.g., Chloramphenicol acetyl transferase, CAT mRNA).
- In Vitro Selection Cycle:
- Reaction: Incubate the ribozyme pool with the target substrate RNA under near-physiological conditions (e.g., low Mg²⁺ concentration, physiological temperature).
- Recovery: Use RT-PCR with primers specific to successful trans-splicing products (fused 5'-substrate and 3'-ribozyme exon) to selectively amplify the functional ribozyme sequences.
- Amplification: Amplify the recovered sequences by PCR for the next round.
- Stringency Adjustment:
- In early selection rounds, use long reaction times and high substrate concentrations to ensure recovery of even weakly functional sequences, preventing their loss from the pool.
- In later rounds, increase stringency by reducing reaction time or substrate concentration to favor the enrichment of ribozymes with the fastest kinetics and highest specificity, thereby minimizing crosstalk [41] [7].
- Validation: Sequence the final pool to identify dominant ribozyme variants and characterize their splicing efficiency and specificity against both the target and off-target substrates.
Visualization of Orthogonal Ribozyme System Design
The following diagrams illustrate the core strategies for achieving orthogonality in trans-splicing ribozyme systems.
Diagram Title: Input-Dependent Ribozyme Assembly for Orthogonal Output
Diagram Title: Ribozyme Crosstalk from Off-Target Interactions
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Developing Orthogonal Ribozyme Systems
| Reagent / Tool | Function | Example & Notes |
|---|---|---|
| Group I Intron Ribozymes | The catalytic RNA core for trans-splicing. | Tetrahymena thermophila (robust, well-characterized) [31] and Azoarcus (small, fast-folding) [7] ribozymes are common starting points. |
| Reporter Systems | Quantitative measurement of splicing efficiency and crosstalk. | Plasmids with ribozyme inserted into sfGFP or eYFP coding sequence [42]. A reference promoter (e.g., pR) expressing eCFP normalizes for extrinsic variation [43]. |
| High-Throughput Screening Platform | Identification of orthogonal variants from large libraries. | Fluorescence-Activated Cell Sorting (FACS) coupled with Next-Generation Sequencing (NGS) [42]. |
| Inhibitor RNA / Toehold Switch | Controls split ribozyme assembly for screening orthogonality. | An RNA molecule that binds one split fragment's guide sequence, preventing assembly until displaced by the target RNA input [42]. |
| Mathematical Model | Guides design and predicts system performance. | Thermodynamic models of RNA-RNA hybridization energy help design specific IGS-substrate pairs and predict crosstalk [42]. Equilibrium models can also predict optimal ribozyme expression levels [43]. |
The journey of a macromolecule within a cell is markedly different from its behavior in dilute buffer solutions. The cellular interior is a densely crowded, compartmentalized, and sticky environment that presents unique challenges for the folding, stability, and delivery of biologics like trans-splicing group I intron ribozymes [44]. For researchers developing these ribozymes for biocomputing applications, understanding these in vivo parameters is crucial for designing systems that function reliably in living cells. Key factors that differentiate the in-cell environment include macromolecular crowding, which can occupy up to 30% of cellular volume, hindered diffusion that affects molecular mobility, vectorial synthesis during translation, and the constant activity of molecular chaperones that assist folding [44]. These factors collectively influence the energy landscape of folding reactions, often in ways that are not replicated in standard in vitro experiments. This application note provides practical methodologies to address these challenges, with specific focus on trans-splicing group I introns as functional components in biological computing systems.
Quantitative Analysis of Cellular Environments
Table 1: Key Environmental Factors Affecting Macromolecular Folding and Stability In Vivo
| Environmental Factor | In Vitro Conditions | In Vivo Conditions | Impact on Folding/Stability |
|---|---|---|---|
| Macromolecular Crowding | Dilute solutions (1-10 mg/ml) | Highly crowded (≈300-400 mg/ml) | Favors compaction; modest effects on native state stability [44] |
| Translational Diffusion | Unhindered | Significantly reduced for proteins >27 kDa [44] | Slows folding kinetics; may affect assembly |
| Rotational Diffusion | Unhindered | Slower with protein crowders vs. inert polymers [44] | Affects conformational sampling during folding |
| Vectorial Synthesis | Full length protein/RNA | N-to-C-terminal emergence from ribosome [44] | Enables co-translational folding of domains |
| Chaperone Assistance | Absent | Present (GroEL/ES, Hsp70, etc.) [44] | Remodels folding energy landscape; prevents aggregation |
Research Reagent Solutions for In Vivo Studies
Table 2: Essential Research Reagents for Studying In Vivo Folding and Delivery
| Reagent/Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| Fluorescent Reporters | FlAsH/ReAsH (bis-arsenical dyes) [45] | Site-specific labeling of tetracysteine-tagged proteins; reports conformational state | Minimal structural perturbation; specific to engineered motifs |
| Chemical Denaturants | Urea (cell-permeant) [45] | In-cell stability measurements via titration | Requires fresh preparation and concentration verification |
| Expression Systems | High-copy number plasmids (pET series) [45] | High-yield expression of target proteins/ribozymes | Choice of E. coli strain (BL21(DE3), WG710, WG708) affects results |
| Delivery Carriers | Lipid nanoparticles, Cell-penetrating peptides [46] | Intracellular delivery of nucleic acids and proteins | Efficiency varies by cell type; potential cytotoxicity |
| Physical Delivery Tools | Electroporation, Microinjection, Sonoporation [47] | Membrane disruption for cargo entry | Balance between efficiency and cell viability critical |
| Ribozyme Variants | Tetrahymena thermophila, Azoarcus group I introns [31] [7] | trans-splicing activity for RNA modification | Azoarcus ribozyme is smaller but less efficient in cells [7] |
Methodological Protocols
Direct Measurement of Protein Stability in Live Cells
The following protocol adapts the FlAsH-based methodology for measuring thermodynamic stability of proteins directly in Escherichia coli cells, which can be extended to study ribozyme-associated proteins or protein-assisted ribozyme folding [45].
Reagents and Equipment
- Plasmid: Target gene cloned into pET16b (or similar) with engineered tetracysteine motif (CCGPCC)
- Cell Line: E. coli BL21(DE3) or equivalent
- Labeling Reagent: FlAsH-EDT₂ (commercially available as Lumio, 2 mM stock in DMSO)
- Denaturant: Urea stock (9 M in 10 mM Tris HCl, pH 7.5, freshly prepared and filter-sterilized)
- Inducer: IPTG (400 mM stock, filter-sterilized)
- Culture Medium: LB with appropriate antibiotics
- Equipment: Fluorescence microscope or plate reader, cell culture apparatus
Expression and Labeling Protocol
- Transform E. coli BL21(DE3) with plasmid encoding tetra-Cys tagged target protein and plate on selective medium.
- Inoculate a single colony into 5 mL LB medium with antibiotic; grow overnight at 37°C with shaking.
- Dilute overnight culture 1:100 into fresh medium with antibiotic; grow at 37°C to OD₆₀₀ ≈ 0.6.
- Induce expression with 0.4 mM IPTG; continue growth for 1-2 hours (optimize for sufficient expression).
- Add FlAsH-EDT₂ to final concentration of 0.5-1 μM; incubate 30-60 minutes with gentle shaking.
- Harvest cells by gentle centrifugation (3000 × g, 5 min); wash twice with buffer to remove excess dye.
In-Cell Urea Titration and Data Analysis
- Prepare urea solutions ranging from 0 to 8 M in appropriate buffer (e.g., 10 mM Tris HCl, pH 7.5).
- Resuspend labeled cells in each urea concentration; incubate 30-60 minutes to reach equilibrium.
- Measure fluorescence emission at ≈535 nm (excitation ≈508 nm) for each sample.
- Determine fraction unfolded at each urea concentration using the relationship: Fracₜᵢₒₙ Uₙfₒₗdₑd = (F - Fₙ)/(Fᵤ - Fₙ), where F is measured fluorescence, Fₙ is native state fluorescence, and Fᵤ is unfolded state fluorescence.
- Plot fraction unfolded versus urea concentration and fit to standard unfolding models to extract ΔG°ᴜₙf.
Critical Considerations
- Motif Placement: Engineer tetracysteine motif in regions that undergo conformational changes during folding/unfolding.
- Labeling Specificity: Include control with untagged protein to assess nonspecific labeling.
- Cell Viability: Verify >90% viability after urea treatment using plating or vital stains.
- Expression Level: Optimize expression time to avoid aggregation; use low-copy plasmids if needed.
Intracellular Delivery Methods for Ribozymes
Efficient intracellular delivery is essential for deploying trans-splicing ribozymes in biocomputing applications. The following protocol compares physical methods suitable for ribozyme delivery.
Microinjection for Single-Cell Delivery
- Principle: Direct mechanical insertion of cargo via glass micropipette [47] [46]
- Procedure:
- Prepare ribozyme solution (0.1-1 mg/mL in suitable buffer).
- Load into glass micropipette (tip diameter 0.1-0.5 μm).
- Immobilize cells on coated coverslip.
- Using micromanipulators, position pipette tip near cell membrane.
- Apply brief pressure pulse (10-100 ms) to deliver 0.1-1 pL volume.
- Retract pipette carefully and assess cell viability.
- Throughput: 100-1500 cells/hour depending on automation level
- Advantages: Direct cytoplasmic or nuclear delivery; precise dosage control
- Limitations: Low throughput; requires specialized equipment and technical skill
Electroporation for Population-Level Delivery
- Principle: Transient membrane permeabilization by electrical pulses [46]
- Procedure:
- Harvest and wash cells; resuspend in electroporation buffer at 10⁷-10⁸ cells/mL.
- Mix cell suspension with ribozyme (10-50 μg/mL final concentration).
- Transfer to electroporation cuvette (0.1-0.4 cm gap).
- Apply optimized electrical pulse (typically 500-1500 V/cm, 1-10 ms pulse length).
- Immediately transfer cells to recovery medium; incubate 10-60 minutes before plating.
- Assess delivery efficiency and cell viability.
- Throughput: 10⁶-10⁸ cells per treatment
- Advantages: High efficiency for nucleic acids; applicable to various cell types
- Limitations: Significant cell death if parameters not optimized; requires ribozyme stability verification
Trans-Splicing Group I Intron Engineering
Ribozyme Designs for Biocomputing
Group I intron ribozymes can be engineered as precise RNA modification tools for biological computing systems. Five principal designs enable different computational operations through RNA sequence manipulation [31]:
Selection Guidelines for Ribozyme Variants
Table 3: Comparison of Group I Intron Ribozymes for Biocomputing Applications
| Ribozyme Source | Size (nt) | Folding Kinetics | Trans-Splicing Efficiency | Optimal Design Context | Ideal Application |
|---|---|---|---|---|---|
| Tetrahymena thermophila [31] [7] | ≈400 | Moderate | High in cells | Extended Guide Sequence (EGS) | High-fidelity message processing |
| Azoarcus [7] | ≈205 | Fast | Moderate in vitro, low in cells | Natural tRNA anticodon stem-loop | Rapid response systems |
| Pneumocystis carinii [31] | ≈300 | Moderate | High with P10/P9.0 elongation | Extended P9.0 helix | 5' replacement operations |
| Didymium/Fuligo [31] | ≈350 | Moderate | Moderate with EGS | Extended Guide Sequence | Specialized substrate recognition |
Experimental Evolution Protocol for Ribozyme Optimization
This protocol describes a method to improve trans-splicing efficiency of group I intron ribozymes through experimental evolution in bacterial cells [31].
Library Construction and Selection
- Randomized Library: Introduce degeneracy in the substrate recognition regions (P1 helix or EGS).
- Selection System: Clone library into vector with antibiotic resistance gene whose activity depends on successful trans-splicing.
- Transformation: Transform E. coli with library; plate on selective medium containing antibiotic.
- Harvest Survivors: Collect resistant colonies after 24-48 hours; isolate plasmids.
- Iterative Rounds: Repeat process for 3-6 rounds to enrich functional variants.
Screening and Characterization
- High-Throughput Assay: Screen individual clones using fluorescence reporters or colorimetric assays.
- Sequence Analysis: Identify consensus sequences and mutations in improved variants.
- Kinetic Analysis: Measure trans-splicing rates for top performers under near-physiological conditions.
- Specificity Testing: Verify minimal off-target splicing using transcriptome-wide analysis.
Visualization of Experimental Workflows
Integrated Data Analysis and Implementation Framework
Successful implementation of trans-splicing ribozymes for biocomputing requires careful consideration of the interrelationships between folding, stability, and delivery parameters. The following framework provides guidance for troubleshooting common challenges:
When low splicing efficiency is observed:
- Verify ribozyme stability using FlAsH-based method in relevant cellular compartment
- Assess delivery efficiency using control fluorescently-labeled ribozymes
- Optimize recognition helix length and GC content for target accessibility
For cell-type specific optimization:
- Prioritize delivery methods based on cell hardness and sensitivity (electroporation for robust cells, microinjection for sensitive primary cells)
- Adapt ribozyme expression levels to minimize cellular stress while maintaining function
- Consider compartment-specific crowding effects when predicting folding kinetics
To enhance computational reliability:
- Implement redundant ribozyme systems with different recognition sequences for critical operations
- Characterize off-target effects in the specific cellular environment
- Establish quantitative input-output relationships for predictable circuit behavior
This application note provides the foundational methodologies for addressing the central challenges in deploying trans-splicing group I intron ribozymes for biocomputing research. By implementing these protocols and considering the integrated framework, researchers can significantly improve the reliability and performance of biological computing systems in cellular environments.
Combinatorial and Machine Learning Approaches for Ribozyme Optimization
Group I intron ribozymes are catalytic RNAs that excise themselves from primary transcripts without requiring the spliceosome, instead catalyzing two consecutive transesterification reactions to remove themselves and ligate flanking exons [48]. These natural cis-splicing ribozymes can be engineered into trans-splicing variants that recognize substrate RNAs through base-pairing interactions, enabling them to replace either the 5' or 3' portion of a substrate RNA with the ribozyme's own exon sequences [48] [7]. This capacity for precise RNA sequence modification makes trans-splicing group I introns particularly valuable for biocomputing research, where they can function as programmable molecular components for implementing logical operations, signal processing, and state transitions within biological systems.
The optimization of these ribozymes for reliable performance in synthetic biological circuits presents substantial challenges, including improving catalytic efficiency, specificity, and stability under physiological conditions. This article details how combinatorial methods and machine learning (ML) approaches are being deployed to overcome these limitations, providing researchers with structured experimental protocols and computational frameworks to advance ribozyme engineering for biocomputing applications.
Ribozyme Engineering Fundamentals: Design Principles and Variations
trans-Splicing Mechanism and Design Topologies
Trans-splicing group I intron ribozymes recognize their target sites on substrate RNAs through specific base-pairing interactions. Five distinct design architectures have been established, which differ primarily in how the splice sites are recognized and which portion of the substrate RNA is modified [48]:
- Type A (5'-Splice Site Recognition): The ribozyme binds the substrate via the P1 helix, leading to replacement of the substrate's 3'-portion with the ribozyme's 3'-exon [48].
- Type B (3'-Splice Site Recognition via P10/P9.0): The ribozyme utilizes P10 and P9.0 helices to bind the substrate, resulting in replacement of the substrate's 5'-portion with the ribozyme's 5'-exon [48].
- Type C (Dual Splice Site Recognition): Combining 3'-splice site recognition (P9.0/P10) with 5'-splice site recognition (P1) enables excision of internal sequences from the substrate RNA [48].
- Type D (3'-Splice Site Recognition via P9.2): The ribozyme employs P9.0 and P9.2 helices for substrate binding, replacing the substrate's 5'-portion with its 5'-exon [48].
- Type E (Spliceozymes): Utilizing both P9.2 (3'-splice site) and P1 (5'-splice site) recognition enables removal of introns up to 100 nucleotides from RNA substrates [48].
The following diagram illustrates the key trans-splicing designs and their molecular interactions:
Comparative Analysis of Model Ribozyme Systems
The selection of an appropriate parent ribozyme constitutes a critical initial step in engineering optimized trans-splicing systems. The table below summarizes key characteristics of the most widely used group I intron ribozymes:
Table 1: Comparison of Model Group I Intron Ribozymes for trans-Splicing Applications
| Ribozyme Source | Length (nt) | Group Classification | Folding Kinetics | trans-Splicing Efficiency | Key Applications |
|---|---|---|---|---|---|
| Tetrahymena thermophila | ~400 | IC1 | Slower | High in vitro and in cells [48] [7] | mRNA repair, evolutionary studies [48] |
| Azoarcus BH72 | 205 | IC3 | Faster [7] | High in vitro, low in cells [7] | Structural studies, mechanistic analysis [23] [7] |
| Pneumocystis carinii | ~300 | IE | Intermediate | Moderate [48] | 3' splice site recognition designs [48] |
Combinatorial Optimization Strategies
High-Throughput Screening with PERSIST-seq
The PERSIST-seq (Pooled Evaluation of mRNA In-solution Stability, and In-cell Stability and Translation RNA-seq) platform enables systematic optimization of ribozyme performance by simultaneously measuring multiple RNA performance parameters [49]. This method is readily adaptable for screening ribozyme libraries by substituting the reporter open reading frame with ribozyme sequences.
Table 2: Key Design Parameters for Ribozyme Optimization via Combinatorial Approaches
| Parameter | Combinatorial Strategy | Impact on Ribozyme Performance |
|---|---|---|
| UTR Variants | Library of 5' and 3' UTRs from viral and human genomes [49] | Enhanced ribosome loading and cellular stability [49] |
| Coding Sequence Structure | "Superfolder" designs with optimized secondary structure [49] | Simultaneous improvement of stability and expression [49] |
| Nucleoside Modifications | Incorporation of pseudouridine (ψ) and derivatives [49] | Enhanced solution stability and reduced immunogenicity [49] |
| Substrate Recognition Helices | Randomized internal guide sequences [7] | Expanded target range and specificity [48] [7] |
Protocol 3.1: PERSIST-seq for Ribozyme Library Screening
Library Design and Synthesis:
- Design ribozyme variants with diversified UTRs, substrate recognition sequences, and nucleoside modifications
- Include unique molecular barcodes (12-15 nt) in the 3' UTR for multiplexed analysis
- Obtain full-length DNA templates through commercial gene synthesis services
In Vitro Transcription and Processing:
- Perform pooled in vitro transcription with T7 RNA polymerase
- Add 5' m⁷G cap and 3' poly(A) tail using enzymatic methods
- Purify RNA library using silica membrane columns
Stability and Expression Profiling:
- Divide library for parallel assays:
- In-cell stability: Transfert mammalian cells (HEK293T), harvest at timepoints (0, 2, 4, 8, 12h)
- In-solution stability: Incubate in physiological buffer at 37°C, sample at identical intervals
- Translation efficiency: Perform polysome profiling on sucrose density gradients
- Extract RNA and convert to cDNA with barcode-specific primers
- Divide library for parallel assays:
Sequencing and Data Analysis:
- Amplify barcode regions with Illumina adapters
- Sequence on high-throughput platform (NovaSeq 6000)
- Calculate degradation rates from timecourse data
- Determine ribosome loading from polysome fractions
- Correlate sequence features with performance metrics
Generative Models for Exploring Ribozyme Sequence Space
Generative models enable exploration of the vast sequence space of functional ribozymes beyond the limitations of traditional mutagenesis approaches. The following workflow illustrates the implementation of Direct Coupling Analysis (DCA) for ribozyme diversification:
Protocol 3.2: Generative Model-Guided Ribozyme Diversification
Training Data Curation:
- Compile multiple sequence alignment (MSA) of group I introns with conserved domain architecture
- Include 800+ sequences from diverse organisms [23]
- Filter for sequences sharing secondary structure features with target ribozyme
Model Training and Validation:
- Implement Direct Coupling Analysis using pseudolikelihood maximization
- Extract coupling parameters Jᵢⱼ and fields hᵢ representing evolutionary constraints
- Validate model by recapitulating known functional sequences
Sequence Generation:
- Sample novel sequences using Markov Chain Monte Carlo sampling
- Target specific mutational distances from reference sequence (10-150 mutations)
- Generate 150+ sequences per mutational bin for comprehensive coverage
Experimental Validation:
- Synthesize library and clone into expression vectors
- Assess self-splicing activity using high-throughput sequencing assay
- Calculate activity scores as log(fraction active after screening/fraction before incubation)
- Set significance threshold at z-score > 3.09 (p < 0.001) [23]
This approach has demonstrated remarkable success, with DCA-generated sequences maintaining activity at significantly higher mutational distances (L₅₀ = 20 mutations, Lmax = 60 mutations) compared to random mutagenesis (L₅₀ = 5 mutations, Lmax = 10 mutations) [23].
Machine Learning Approaches for Ribozyme Design
Dataset Curation and Feature Engineering
The development of effective ML models for ribozyme design requires large, high-quality training datasets. Recent efforts have created comprehensive resources containing over 320,000 RNA structures with lengths ranging from 5 to 3,538 nucleotides [50]. These datasets specifically emphasize complex structural motifs, including multi-branched loops and n-way junctions that present particular challenges for ribozyme engineering.
Table 3: Machine Learning Models for Ribozyme Optimization
| Model Class | Representative Algorithms | Application in Ribozyme Engineering | Performance Considerations |
|---|---|---|---|
| Generative Models | DCA [23], RiboDiffusion [50] | Exploring neutral network of self-reproducing sequences | DCA achieves Lmax = 60 mutations from reference [23] |
| Inverse Folding | RNAinverse, INFO-RNA, Meta-LEARNA [50] | Designing sequences for target secondary structures | Accuracy varies with structure complexity and length [50] |
| Stability Prediction | DegScore [49], PERSIST-seq models [49] | Predicting in-solution and cellular RNA half-life | Enables simultaneous optimization of stability and expression [49] |
Integrated Experimental-Computational Workflow
The most effective ribozyme optimization pipelines combine computational prediction with experimental validation. The following protocol outlines an iterative design-build-test cycle for ribozyme engineering:
Protocol 4.2: Iterative Ribozyme Optimization Pipeline
Initial Sequence Design:
Library Construction and Screening:
- Synthesize variant library with strategic diversification of key functional domains
- Clone into appropriate expression vectors
- Measure splicing efficiency in vitro under near-physiological conditions (2mM Mg²⁺, 37°C) [7]
Model Refinement:
- Train ML models on experimental activity data
- Incorporate additional features (structural accessibility, thermodynamic parameters)
- Generate improved designs using updated models
Validation in Biocomputing Applications:
- Test optimized ribozymes in intended biocomputing context
- Assess orthogonality in multi-component systems
- Evaluate performance over multiple operational cycles
Table 4: Key Research Reagent Solutions for Ribozyme Engineering
| Reagent/Resource | Specifications | Application | Example Sources |
|---|---|---|---|
| Group I Intron Templates | Tetrahymena thermophila (400 nt), Azoarcus (205 nt) [48] [7] | Baseline ribozyme constructs | AddGene, scientific literature |
| High-Throughput Synthesis | Pooled DNA libraries (10³-10⁵ variants) [49] | Combinatorial library generation | Twist Bioscience, GenScript |
| In Vitro Transcription Kit | T7 RNA polymerase, modified NTPs (pseudouridine) [49] | Ribozyme production | ThermoFisher, NEB |
| Stability Assessment Platform | PERSIST-seq protocol [49] | Simultaneous stability and translation measurement | Custom implementation |
| Generative Model Code | DCA implementation for RNA [23] | Exploring ribozyme sequence space | GitHub repositories |
| Standardized Dataset | 320,000+ RNA structures [50] | Training ML models | RNAsolo, Rfam databases |
Application Notes for Biocomputing Implementation
Practical Considerations for System Integration
When implementing optimized trans-splicing ribozymes in biocomputing systems, several practical considerations emerge from experimental studies:
- Cellular Environment Effects: Ribozymes demonstrating high activity in vitro may show significantly reduced efficiency in cellular environments. The Azoarcus ribozyme, for instance, exhibits robust in vitro activity but poor performance in E. coli cells [7].
- Structure-Function Tradeoffs: Highly structured "superfolder" designs can improve stability but may impede ribosomal access during translation initiation [49].
- Orthogonality Requirements: In multi-component biocomputing systems, ribozyme-substrate interactions must be engineered to minimize cross-talk through careful design of recognition helices.
Troubleshooting Common Experimental Challenges
- Low Splicing Efficiency: Enhance substrate accessibility by extending guide sequences (EGS) and optimizing P1 helix length (typically 6-8 bp) [7].
- Poor Cellular Stability: Incorporate stabilizing 3' UTR elements (e.g., MALAT1 ENE) and implement nucleoside modifications (pseudouridine) [49].
- Limited Target Range: Utilize generative models (DCA) to explore sequence space beyond natural ribozyme diversity [23].
- Unexpected Splicing Products: Validate specificity through rigorous RNA-seq analysis and implement additional targeting constraints.
The integration of combinatorial optimization and machine learning approaches provides a powerful framework for advancing ribozyme engineering. By implementing these detailed protocols and leveraging the curated resources outlined in this article, researchers can systematically develop enhanced trans-splicing group I intron ribozymes for sophisticated biocomputing applications.
Benchmarking and Validation: Assessing Ribozyme Efficacy Across Systems and Models
Group I intron ribozymes, which catalyze their own excision from RNA transcripts and ligation of flanking exons, have emerged as powerful and programmable platforms for synthetic biology [25]. Their ability to be converted from cis- to trans-splicing configurations enables the re-writing of genetic information within a cell, a property that is being harnessed for complex cellular logic computation [25]. Among the thousands of known group I introns, the ribozymes from Tetrahymena thermophila and the bacterium Azoarcus represent two of the most well-characterized and functionally distinct systems. This application note provides a comparative analysis of their structural and functional characteristics, along with detailed protocols for their application in trans-splicing and genetic circuit design, framed within the context of advancing biocomputing research.
Comparative Ribozyme Analysis: Structural and Functional Properties
The Tetrahymena and Azoarcus ribozymes differ significantly in their architectural and functional properties, which informs their selection for specific applications. The table below summarizes their key characteristics.
Table 1: Comparative Analysis of Tetrahymena thermophila and Azoarcus Ribozymes
| Characteristic | Tetrahymena thermophila Ribozyme | Azoarcus Ribozyme |
|---|---|---|
| Origin | Eukaryotic, nuclear rRNA [51] | Bacterial, tRNAIle [7] |
| Size | ~400 nucleotides (L-21 ScaI variant) [51] | ~200 nucleotides [7] |
| Folding Kinetics | Slower folding in vitro [7] | Rapid folding in vitro [7] |
| Core Domain Structure | Conserved P3-P7 and P4-P6 domains; Peripheral regions stabilize core [52] | Highly compact core; "Pseudoknot belt" structure [53] |
| Catalytic Activity In Vitro | High trans-splicing efficiency [7] | High activity, comparable to Tetrahymena with optimized design [7] |
| Catalytic Activity In Vivo | Demonstrated mRNA repair in vivo [7] | Significantly lower activity in E. coli cells compared to Tetrahymena [7] |
| Response to Molecular Crowding | Not specifically reported in results | Activity increased at physiological Mg2+; stabilized native state [54] |
| Key Structural Feature | Pre-organized active site in P3-P9 domain [51] | Extensive base stacking; >90% of possible stacking interactions observed [53] |
Application in Trans-Splicing and Biocomputing
A key engineering principle is the conversion of these ribozymes from cis to trans-splicing configurations by splitting the intron and utilizing External Guide Sequences (EGS) to programmatically target specific mRNA substrates [25]. This forms the basis of the SENTR (Split-intron-Enabled RNA Trans-splicing Riboregulator) system.
Table 2: Trans-Splicing Application Notes for Biocomputing
| Application Parameter | Tetrahymena Ribozyme | Azoarcus Ribozyme |
|---|---|---|
| EGS Design Principle | 5'-terminal extended guide sequence (EGS) [7] | EGS that mimics natural cis-splicing context (e.g., base-pairing with 3' exon) [7] |
| Splice Site Recognition | Binds substrate via P1 helix; requires U at splice site paired to G in IGS [7] | Favors the same splice sites as Tetrahymena ribozyme on a given substrate [7] |
| Orthogonality | Can be engineered for orthogonal splicing pathways [25] | Can be engineered for orthogonal splicing pathways with de novo EGS design [25] |
| Logic Gate Implementation | Used in layered genetic circuit designs | Enables complex, single-layer computation (e.g., 6-input AND gates) when coupled with protein splicing [25] |
Experimental Protocols
Protocol: In Vitro Trans-Splicing Activity Assay for Azoarcus Ribozyme
This protocol measures the single-turnover cleavage rate of the Azoarcus ribozyme, which reports on the fraction of natively folded ribozyme and can be adapted for Tetrahymena [54].
I. Materials and Reagents
- Purified L-3 Azoarcus ribozyme (201 nt): The catalytic RNA.
- 32P-labeled RNA substrate (5'-rCAUAUCGCC): Traceable cleavage target.
- GTP (0.5 mM): Nucleophile for the transesterification reaction.
- Reaction Buffer: 20 mM Tris-HCl, pH 7.5.
- MgCl2 Stock Solution: Varying concentrations (1-20 mM).
- Crowding Agents (Optional): e.g., PEG 1000 or Ficoll.
- Stop Solution: Formamide with EDTA.
II. Procedure
- Ribozyme Folding: Denature the ribozyme (0.4 mg/mL in reaction buffer) at 50°C for 5 minutes. Pre-equilibrate at 37°C for 30 minutes in the presence of the desired concentration of MgCl2 and any crowding agents [54].
- Initiate Reaction: Add the 32P-labeled substrate RNA (final concentration ~1 nM) and GTP (0.5 mM) to the pre-folded ribozyme (final concentration 6 µM) [54].
- Time-course Sampling: Withdraw aliquots from the reaction mixture at time intervals (e.g., 25 seconds to 30 minutes) and immediately mix with stop solution to quench the reaction [54].
- Product Analysis: Separate the cleaved and uncleaved RNA products using denaturing polyacrylamide gel electrophoresis. Quantify the product bands using a phosphorimager.
- Data Fitting: Fit the fraction of cleaved product over time to a biphasic, partially compressed exponential model to extract the amplitude (A, representing the active fraction of ribozyme) and the observed rate constant (k1) for the initial fast phase [54].
Protocol: Programming a SENTR System for Logic Computation
This protocol outlines the steps for using split introns to regulate gene expression via RNA trans-splicing [25].
I. Materials and Reagents
- EGS-Intron Fusion Constructs: Plasmid DNA encoding the 5' and 3' intron halves, each fused to computationally designed External Guide Sequences (EGS).
- Target Gene Construct: Plasmid containing the gene of interest (e.g., fluorescent protein, transcription factor) interrupted by the target sequence for the EGSs.
- Host Cells: E. coli or other appropriate microbial hosts.
II. Procedure
- EGS Design: Using machine learning models or sequence design rules, design EGSs complementary to the target mRNA region. For orthogonality, ensure EGS pairs have low sequence similarity [25].
- Genetic Construct Assembly: Clone the EGS sequences, fused to the split intron fragments, into an expression vector. Assemble the target gene construct where the EGS-binding sites flank the sequence to be spliced out.
- Transformation and Expression: Co-transform the EGS-intron and target gene plasmids into host cells.
- Logic Gate Execution: Input signals (e.g., inducing small RNAs) trigger the assembly of the split intron via EGS hybridization. The reconstituted intron performs trans-splicing, excising the target sequence and ligating the exons to produce a functional output mRNA (e.g., for a transcription factor) [25].
- Output Measurement: Assay for the functional output, such as fluorescence for reporter genes or activation of a downstream promoter by the spliced transcription factor.
Essential Research Reagent Solutions
Table 3: Key Reagents for Ribozyme-Based Biocomputing
| Research Reagent | Function and Application |
|---|---|
| L-16 ScaI Tetrahymena Ribozyme | Full-length ribozyme variant for structural studies of splicing intermediates; contains extended Internal Guide Sequence (IGS) for forming dynamic P1 and P10 helices [51]. |
| L-3 Azoarcus Ribozyme | Shortened, highly active variant for in vitro biochemical and biophysical assays; includes sequence for oligonucleotide substrate binding [54]. |
| External Guide Sequences (EGS) | De-novo-designed RNA guides programmed to hybridize with target mRNAs and control the assembly and activity of split introns for trans-splicing [25]. |
| Orthogonal Split Intein Pairs | Protein splicing elements used in conjunction with split introns to create multi-input logic gates by assembling a single functional protein from multiple peptides [25]. |
| Molecular Crowders (PEG, Ficoll) | Macromolecular agents used in vitro to mimic intracellular crowded conditions, which stabilize the native ribozyme structure and enhance catalytic activity at physiological Mg2+ levels [54]. |
Visualization of Concepts and Workflows
SENTR System Workflow
Six-Input AND Gate Logic
The precise manipulation of genetic information processing is a foundational goal in synthetic biology and biocomputing. Among the various molecular tools available, group I introns represent a powerful class of self-splicing ribozymes that catalyze their own excision from precursor RNA molecules through two consecutive transesterification reactions [2]. These catalytic RNAs are characterized by highly conserved core structures that facilitate precise exon ligation, making them ideal candidates for engineering programmable genetic circuits [2]. Recent advances have demonstrated that group I introns can be harnessed not only for conventional cis-splicing but also for novel trans-splicing applications that enable the reconstruction of functional RNAs from separate transcripts. This capability is particularly valuable for biocomputing systems that require conditional activation or logical operations based on multiple molecular inputs. The development of techniques such as PIET (Permuted Intron-Exon through Trans-splicing) and CIRC (Complete self-splicing Intron for RNA Circularization) has significantly expanded the toolbox available for RNA-based circuit design [9]. This Application Note provides detailed protocols for validating group I intron-based splicing systems across increasingly complex biological environments, from controlled in vitro reactions to prokaryotic and eukaryotic cellular models, with specific emphasis on their integration into biocomputing architectures.
In Vitro Splicing Assays: Foundation for Validation
High-Throughput Splicing Quantification Using EJIPT
The Exon Junction Complex Immunoprecipitation (EJIPT) assay provides a robust platform for quantitative analysis of splicing efficiency by detecting the unique molecular signature that splicing leaves on mRNAs [55]. When splicing occurs successfully, it deposits an Exon Junction Complex (EJC) approximately 20-24 nucleotides upstream of splice junctions, with core components including eIF4AIII and Y14 proteins [55]. This assay is particularly valuable for biocomputing applications as it enables rapid screening of multiple intron designs under various conditions.
Table 1: Key Reagents for EJIPT Splicing Assay
| Component | Specifications | Function in Assay |
|---|---|---|
| Biotin-labeled pre-mRNA | Adenovirus type 2 construct (Ad2ΔIVS), 40 nM in reaction, ~3 biotin molecules/RNA | Splicing substrate for capture and quantification |
| Splicing Extract | HEK 293T whole-cell extract, 80 μg per 20 μl reaction | Provides cellular machinery for splicing reaction |
| Coated Plates | Black-well NeutrAvidin-coated plates (Pierce) | Immobilizes biotinylated RNA complexes |
| Primary Antibody | Anti-eIF4AIII (3F1) antibody, 1:350 dilution in HNT buffer | Specifically binds EJC component on spliced mRNA |
| Detection System | HRP-conjugated anti-mouse IgG + Super Signal ELISA Fempto substrate | Generates chemiluminescent signal for quantification |
Protocol: High-Throughput EJIPT in 384-Well Format
Reaction Assembly: Using a liquid handling robot (e.g., Beckman Coulter Biomek FX), assemble 20-μl splicing reactions in 384-well plates containing:
- 1× SP buffer (0.5 mM ATP, 20 mM creatine phosphate, 1.6 mM MgCl₂)
- 80 μg whole-cell splicing extract
- 40 nM biotin-labeled Ad2ΔIVS pre-mRNA
- 1 U/μl RNasin RNase inhibitor
- Test compounds (optional, for inhibitor screening) in 1% DMSO final concentration
Splicing Reaction: Incubate plates for 1.5 hours at 30°C to allow complete splicing.
Complex Capture: Dilute reactions with 40 μl HNT buffer (20 mM HEPES-KOH pH 7.9, 150 mM NaCl, 0.5% Triton X-100) and transfer 50 μl to NeutrAvidin-coated plates. Incubate 1 hour at room temperature.
Wash Steps: Aspirate samples using a microplate washer (e.g., Bio-Tek ELx405) with six wash cycles using HNT buffer.
EJC Detection:
- Add 40 μl primary anti-eIF4AIII antibody (1:350 in HNT with 1 μg/ml BSA)
- Incubate 1 hour at room temperature
- Wash plates as in step 4
- Add HRP-conjugated anti-mouse IgG secondary antibody (1:10,000 in HNT)
- Incubate 1 hour at room temperature
- Perform final wash series
Signal Detection: Add 50 μl Super Signal ELISA Fempto chemiluminescent substrate and measure luminescence on a plate reader (e.g., Perkin Elmer Envision) [55].
Magnetic Bead-Based Splicing Validation
For lower-throughput validation studies or when analyzing multiple time points, a magnetic bead-based approach offers flexibility with reliable quantification.
Protocol: 96-Well Magnetic Bead Assay
Splicing Reaction: Assemble 10-μl reactions on ice in 96-well plates containing:
- 1× SP buffer
- 40 μg splicing extract
- 20 nM biotin-labeled pre-mRNA
- 1 U/μl RNasin
- Incubate 1.5 hours at 30°C
Antibody Immobilization: During splicing incubation, immobilize primary antibodies on protein A magnetic beads (>1 hour at 4°C in PBS-0.1% NP-40) then wash extensively.
Immunoprecipitation: Add 100 μl IP reaction mixtures to each well containing:
- 8.6 μl bead suspension with immobilized antibody
- Primary antibody (0.7-1.7 μl depending on antibody)
- 0.2 U/μl RNasin in HNT buffer
- Perform IP for 1 hour with shaking (750 rpm) at 30°C
Wash and Detection:
- Wash beads five times with 200 μl HNT buffer using magnetic particle processor
- Incubate with 120 μl avidin-HRP conjugate (1:25,000 in HNT) for 1 hour
- Wash five times with 200 μl HNT
- Add 150 μl Super Signal ELISA Fempto substrate for detection [55]
Diagram 1: EJIPT assay workflow for splicing quantification
Advanced RNA Circularization Techniques for Biocomputing
CIRC Method for Efficient RNA Circularization
The Complete self-splicing Intron for RNA Circularization (CIRC) method represents a significant advancement for biocomputing applications requiring stable RNA structures, as circRNAs demonstrate enhanced stability and extended half-life compared to linear RNAs [9]. This technique utilizes intact group I introns, eliminating the need for engineering split introns and streamlining the production of covalently closed RNA circles.
Protocol: CIRC-Based RNA Circularization
Template Design:
- Clone the full-length coding sequence of your RNA of interest between the 5' and 3' exons of a group I intron (e.g., Anabaena)
- Ensure the construct maintains the native intron structure without fragmentation
- Note: Removal of homology arms typically required in PIE methods enhances CIRC efficiency [9]
In Vitro Transcription:
- Use T7 or SP6 RNA polymerase systems for transcription
- Include 2+ guanosine residues at the 5' end for efficient T7 transcription initiation
- Use standard NTP mix (no special modifications required)
- Incubate 2-4 hours at 37°C
Circularization Reaction:
- Assemble 50-μl reactions containing:
- 1-2 μg linear pre-RNA transcript
- 50-100 mM MgCl₂ (concentration optimization recommended)
- Splicing buffer (pH ~7.5, milder conditions than PIE required)
- Note: GTP cofactor not required as CIRC bypasses first transesterification step [9]
- Incubate 1-2 hours at 37-45°C (reduced time compared to PIE)
- Assemble 50-μl reactions containing:
Product Purification:
- Option 1: RNase R treatment to degrade linear RNAs
- Option 2: Oligo(dT)-based purification for polyA- circRNA isolation
- Validate circularization by RT-PCR across junction and RNase R resistance [9]
PIET System for Controlled Trans-Splicing
The Permuted Intron-Exon through Trans-splicing (PIET) method provides a two-component system that enables precise temporal control over circularization initiation, making it particularly valuable for conditional biocomputing operations.
Protocol: PIET Trans-Splicing Circularization
RNA Component Preparation:
- Synthesize two separate RNA fragments via in vitro transcription:
- Fragment A: 5' half-intron with 5' exon
- Fragment B: Intermediate containing 3' intron portion with 3' exon
- Purify using standard RNA cleanup protocols
- Synthesize two separate RNA fragments via in vitro transcription:
Trans-Splicing Reaction:
- Combine components in molar ratios (optimize 5'-intron:intermediate ratio >5:1):
- 5-125 nM Fragment A (5' half-intron)
- 10 nM Fragment B (intermediate)
- Splicing buffer with MgCl₂
- Incubate 1-2 hours at 37°C
- Monitor efficiency by adjusting 5'-intron-to-intermediate ratio [9]
- Combine components in molar ratios (optimize 5'-intron:intermediate ratio >5:1):
Validation and Applications:
- Confirm circRNA formation by:
- RT-PCR demonstrating precise exon ligation
- RNase R resistance assay
- Northern blot analysis
- For biocomputing applications, design split components to respond to different molecular inputs
- Confirm circRNA formation by:
Table 2: Quantitative Comparison of RNA Circularization Methods
| Parameter | PIE Method | PIET Method | CIRC Method |
|---|---|---|---|
| Intron Engineering | Requires specific split sites | Uses split intron components | No intron splitting required |
| Mg²⁺ Requirements | High (often >100 mM) | Moderate | Moderate (50-100 mM) |
| Time Efficiency | Lengthy incubation (2-4 hrs) | Moderate (1-2 hrs) | Rapid (1-2 hrs) |
| GTP Requirement | Essential for first step | Not required | Not required |
| Homology Arm | Required for split intron binding | Required for component interaction | Unnecessary (enhances efficiency) |
| Size Capacity | Limited for large RNAs (<9 kb) | Moderate | Excellent (tested up to 12 kb) |
| Control Options | Single-component system | Two-component temporal control | Single-component simplicity |
Diagram 2: Comparison of CIRC and PIET circularization methods
Cellular Validation in Prokaryotic and Eukaryotic Systems
Fluorescent Reporter Systems for Splicing Validation
The development of fluorescent reporter systems enables quantitative assessment of splicing efficiency in live cells, providing crucial validation for biocomputing circuits before full implementation.
Protocol: Fluorescent Splicing Reporter in T. vaginalis Model
Reporter Construct Design:
- Clone intron of interest between constitutive promoter and GFP coding sequence
- For trans-splicing validation, split intron between two separate constructs
- Include appropriate control constructs (intron-less and mutation controls)
Cell Transfection and Culture:
- Transfert T. vaginalis cells using electroporation (optimized parameters: 350 V, 950 μF)
- Culture transfected cells for 24-48 hours to allow reporter expression
- Include untransfected controls for autofluorescence correction
Analysis and Quantification:
- Monitor fluorescence intensity by flow cytometry or fluorescence microscopy
- Compare with controls to calculate splicing efficiency
- For trans-splicing, verify capability to reassemble split introns from Giardia lamblia [56]
- Assess unusual splicing features including degenerate 5' splice sites and extreme branch point juxtaposition [56]
Validation in Human Cell Models
Protocol: circRNA Functionality Assessment in HEK293T Cells
circRNA Production and Delivery:
- Generate circRNA encoding reporter proteins (e.g., EGFP) using CIRC method
- Incorporate IRES elements for cap-independent translation
- Transfect HEK293T cells using lipid-based transfection reagents
- Use linear RNA counterparts as controls
Functional Assessment:
- Monitor EGFP expression 24-48 hours post-transfection via fluorescence microscopy or flow cytometry
- Compare protein expression levels between circRNA and linear RNA transcripts
- Note: circRNA typically demonstrates higher and more sustained protein expression [9]
Stability Analysis:
- Measure RNA half-life by time-course analysis with transcriptional inhibitors
- Perform RNase R treatment to confirm circular nature of transcripts
- Assess immunogenicity potential by measuring interferon response markers
The Scientist's Toolkit: Essential Research Reagents
Table 3: Research Reagent Solutions for Splicing Validation
| Category | Specific Product/Resource | Application Notes |
|---|---|---|
| Splicing Assay Kits | EJIPT components (custom) | High-throughput screening of splicing modulators |
| Intron Templates | Anabaena group I intron constructs | CIRC and PIE applications; requires T7 promoter adaptation |
| Cell Extracts | HEK 293T whole-cell extracts | Maintain splicing competency; prepare fresh or aliquot frozen |
| Detection Antibodies | Anti-eIF4AIII (3F1 clone) | Mouse monoclonal; EJC component recognition |
| Coated Plates | Black-well NeutrAvidin plates (Pierce) | Minimize background in luminescence detection |
| RNA Polymerases | T7 Megascript kits (Ambion) | High-yield RNA synthesis; requires 5' GG for efficiency |
| Magnetic Beads | Protein A magnetic beads (Invitrogen) | Antibody immobilization for bead-based assays |
| Chemical Inhibitors | BN82685 (Calbiochem) | Second-step splicing inhibitor; controls for 1,4-naphthoquinones |
| Reverse Transcriptases | MMLV-derived RTase | cDNA synthesis for splicing intermediate analysis |
Troubleshooting and Technical Considerations
When implementing these validation protocols, several technical considerations require attention. For in vitro splicing assays, maintaining extract competency is crucial - repeated freeze-thaw cycles significantly reduce splicing efficiency. For CIRC applications, the number of guanosine residues at the 5' end influences RNA yield but not circularization efficiency, requiring optimization of transcription templates [9]. In cellular validation systems, distinguishing between true trans-splicing events and experimental artifacts remains challenging; implementation of rigorous controls including RT-free samples and genomic DNA contamination checks is essential [57]. Recent advances in long-read sequencing technologies (Pacific Biosciences and Oxford Nanopore) provide powerful complementary validation by enabling full-length transcript analysis and direct detection of splicing intermediates [58]. For biocomputing applications specifically, consider implementing the PIET system when temporal control over circuit activation is desired, while the CIRC method provides superior efficiency for stable expression outputs.
In the expanding field of synthetic biology, the programming of cellular functions using engineered genetic circuits is a key frontier. A significant challenge in scaling up these circuits is the limited number of regulatory mechanisms that are highly programmable, efficient, and orthogonal [26]. Trans-splicing group I intron ribozymes have emerged as a powerful platform for post-transcriptional gene regulation and RNA repair. These catalytic RNAs can be engineered to recognize and correct mutant mRNAs by replacing their defective portions with healthy sequences, thereby restoring normal protein function [31] [7].
This application note details the validation of a novel class of these tools—Split-Intron-Enabled Trans-splicing Riboregulators (SENTRs)—for therapeutic mRNA repair in human cell models. We provide a comprehensive protocol for designing, delivering, and quantifying the repair of a disease-relevant mRNA target, framing the process within the context of biocomputing research where precise, multi-input logic controls cellular outcomes [22] [26].
Background and Principle
Naturally occurring group I introns are cis-splicing ribozymes that catalyze their own excision from primary RNA transcripts and ligate the flanking exons without the need for a spliceosome [31] [7]. Engineers have harnessed and repurposed this catalytic activity for trans-splicing, where the ribozyme acts on a separate substrate mRNA molecule.
The core mechanism of the SENTR system involves two key steps:
- Recognition: The ribozyme is designed with an External Guide Sequence (EGS) that base-pairs with a specific target sequence on the mutant mRNA, forming a duplex known as the P1 helix.
- Splicing and Repair: The ribozyme then catalyzes a transesterification reaction, excising the defective portion of the target mRNA and ligating its own encoded "therapeutic exon"—-such as a wild-type coding sequence—-to the remaining upstream exon of the substrate [31] [26]. This process effectively repairs the mRNA at the transcript level.
The following diagram illustrates the core trans-splicing repair mechanism and its integration with biocomputing logic.
Materials and Reagents
Research Reagent Solutions
The following table catalogues the essential materials and reagents required for the implementation of this mRNA repair protocol.
Table 1: Key Research Reagents and Materials
| Item | Function / Description | Example / Source |
|---|---|---|
| SENTR Plasmid DNA | Template for in vitro transcription (IVT) of the riboregulator. Contains the engineered group I intron and therapeutic exon. | Custom design based on Gao et al. [26] |
| Lipid Nanoparticles (LNPs) | Delivery vehicle for efficient transfection of SENTR RNA into human cells. Protects RNA and enhances endosomal escape. | As used in mRNA therapeutics [59] [60] |
| Modified Nucleotides | Incorporation during IVT to enhance RNA stability and reduce immunogenicity. | N1-methylpseudouridine (m1Ψ) [59] |
| CleanCap AG | Co-transcriptional capping analog for IVT mRNA. Improves translation efficiency and reduces innate immune sensing. | Triucleotide cap analog (m7GpppAm) [60] |
| Target Reporter Plasmid | Plasmid expressing the mutant mRNA target, often fused to a fluorescent reporter for easy quantification. | e.g., psfGFP-EGFP Target [26] |
| qPCR Assays | For quantifying the levels of repaired mRNA transcript. | TaqMan or SYBR Green assays |
| Antibodies | For Western blot analysis of the repaired, functional protein. | Target-specific antibodies |
Experimental Protocol
SENTR Riboregulator Design and In Vitro Transcription
- EGS and Therapeutic Exon Design: Design the External Guide Sequence (EGS) to be complementary to a region 6-10 nucleotides upstream of the target splice site (a uridine residue) on the mutant mRNA [31] [7]. Clone the therapeutic exon (e.g., wild-type coding sequence) 3' to the group I intron ribozyme (e.g., from Tetrahymena thermophila) in the plasmid vector.
- Template Preparation: Linearize the purified SENTR plasmid DNA downstream of the poly(A) tail region.
- In Vitro Transcription (IVT): Synthesize the SENTR RNA using a T7 RNA polymerase-based IVT system. To produce high-quality, therapeutic-grade RNA:
- Nucleotide Mix: Use a nucleotide mixture where uridine is fully replaced with N1-methylpseudouridine (m1Ψ) to enhance stability and minimize immune activation [59].
- Capping: Employ the CleanCap AG trinucleotide cap analog co-transcriptionally to achieve >94% Cap-1 structure formation, which is critical for high translation efficiency and low immunogenicity [60].
- Purification: Purify the transcribed SENTR RNA using silica column-based kits or HPLC to remove abortive transcripts, dsRNA contaminants, and unincorporated nucleotides.
Cell Culture and Transfection
- Cell Line: Culture appropriate human cell lines (e.g., HEK-293, HeLa) in recommended media supplemented with fetal bovine serum.
- Co-transfection: Plate cells in a 24-well plate to reach 70-80% confluency at the time of transfection.
- Group 1 (Test): Transfect with a mixture of 200 ng of target reporter plasmid and 400 ng of SENTR RNA complexed with lipid nanoparticles (LNPs).
- Group 2 (Negative Control): Transfect with the target reporter plasmid and a non-functional (scrambled EGS) SENTR RNA.
- Group 3 (Positive Control): Transfect with a plasmid directly encoding the fully repaired, wild-type mRNA.
Validation and Quantification of Repair
The following workflow outlines the key steps for validating mRNA repair, from transfection to final analysis.
- Functional Assay (Flow Cytometry): If the target is a fluorescent reporter (e.g., a mutant sfGFP repaired to EFP), analyze cells 48-72 hours post-transfection using a flow cytometer. Measure the mean fluorescence intensity (MFI) in the relevant channel as a direct indicator of functional protein production.
- Molecular Confirmation (RT-qPCR): Extract total RNA from transfected cells and synthesize cDNA.
- Perform qPCR using a primer pair where the forward primer binds in the upstream exon of the target mRNA and the reverse primer binds uniquely within the therapeutic exon brought by the SENTR. This setup ensures specific amplification of only the successfully trans-spliced product.
- Protein Analysis (Western Blot): Lyse a portion of the transfected cells and subject the proteins to SDS-PAGE and Western blotting. Use an antibody specific to the repaired protein to confirm the presence and size of the functional protein.
Data Analysis and Expected Results
Key Performance Metrics
The efficiency of the SENTR-mediated repair should be evaluated using the following quantitative metrics. The data summarized in the table below represents typical outcomes achievable with optimized systems.
Table 2: Quantitative Metrics for mRNA Repair Efficiency
| Metric | Method of Measurement | Expected Outcome with SENTR | Negative Control |
|---|---|---|---|
| Repair Efficiency | RT-qPCR (ratio of repaired mRNA to total target mRNA) | 10- to 50-fold increase over background [26] | Baseline (1x) |
| Protein Restoration | Flow Cytometry (Mean Fluorescence Intensity) | >80% of cells show fluorescence restoration [26] | <5% of cells |
| Splicing Precision | RNA-Seq / Northern Blot | >99% accurate splice junction [31] | N/A |
| Dynamic Range | Dose-response (Output vs. SENTR concentration) | Low background, high induction (>100-fold) [26] | Minimal response |
Troubleshooting Table
Table 3: Common Experimental Challenges and Solutions
| Problem | Potential Cause | Suggested Solution |
|---|---|---|
| Low Repair Efficiency | Poor target site accessibility; weak EGS binding. | Re-design EGS to target a different, more accessible region of the mRNA. Use RNA folding software to predict open loops. |
| High Background in Controls | Off-target splicing. | Increase the specificity of the EGS by checking for unintended complementarity to other mRNAs. Optimize P1 helix length. |
| Low Cell Viability | Cytotoxicity of transfection reagent or LNP. | Titrate the amount of SENTR RNA and LNP. Use a different, less toxic transfection reagent. |
| No Protein Detected | Ribozyme misfolding; poor catalytic activity. | Verify ribozyme activity in a cell-free system first. Ensure the therapeutic exon has a strong Kozak sequence and is in-frame. |
This application note provides a validated protocol for using engineered trans-splicing group I intron ribozymes, specifically SENTRs, to repair mutant mRNAs in human cell models. The system demonstrates high efficiency, precision, and a wide dynamic range, making it a powerful tool for both therapeutic development and advanced synthetic biology applications [26]. By integrating this RNA-level repair mechanism with protein-splicing elements, such as split inteins, this platform can be further expanded to implement complex multi-input logic gates for sophisticated biocomputing and cellular programming [22]. The future of mRNA repair lies in refining delivery, enhancing orthogonality for multi-gene targeting, and advancing towards preclinical validation of these promising tools.
In the field of synthetic biology and biocomputing, trans-splicing group I intron ribozymes have emerged as powerful tools for engineering genetic circuits and implementing Boolean logic within living cells [31]. These catalytic RNA molecules can be designed to perform precise sequence modifications on separate substrate mRNAs, enabling the construction of complex biological computations [7]. The functional performance of these systems hinges on two critical parameters: the splicing efficiency of the ribozyme itself and the logic gate performance of the resulting genetic circuit. Accurate measurement of these parameters is essential for developing reliable biological computing systems with predictable inputs and outputs. This application note provides detailed methodologies for quantifying these functional readouts, framed within the context of advancing biocomputing research using trans-splicing group I introns.
Quantitative Analysis of Splicing and Gate Performance
Key Performance Metrics and Measurement Techniques
The tables below summarize core metrics and methods for evaluating splicing efficiency and logic gate performance in biocomputing systems.
Table 1: Key Metrics for Assessing Splicing Efficiency
| Metric | Description | Common Measurement Techniques |
|---|---|---|
| Splicing Yield | Percentage of substrate RNA correctly spliced by the ribozyme. | RT-PCR, Primer Extension, Northern Blot [61]. |
| Reaction Kinetics | Rates of the two transesterification reactions ((k1), (k2)). | Stopped-flow assays with radiolabeled substrates [31] [7]. |
| Fidelity/Specificity | Ability to discriminate correct vs. incorrect splice sites. | Deep sequencing of splicing products [62]. |
| In Vivo Splicing Efficiency | Splicing activity within a cellular environment. | Reporter assays (e.g., fluorescence restoration), RT-qPCR [61] [63]. |
Table 2: Parameters for Evaluating Logic Gate Performance
| Parameter | Description | Impact on Circuit Function |
|---|---|---|
| Output Dynamic Range | Ratio between ON and OFF states of the gate. | Determines signal clarity and ability to drive downstream components [63]. |
| Leakiness (Basal Activity) | Output level in the absence of one or more inputs. | Reduces signal-to-noise ratio; can be mitigated by splitting highly active proteins [64]. |
| Response Time | Time delay between input presence and output detection. | Governs computational speed; influenced by splicing kinetics and protein maturation [64] [63]. |
| Fan-out | Number of downstream gates an output can reliably drive. | Critical for scaling circuits to greater complexity [65]. |
Table 3: Computational Tools for Predicting Splice-Disruptive Effects
| Tool Type | Example | Application in Biocomputing |
|---|---|---|
| Deep Learning-Based Models | Not specified in results | Genome-wide annotation of splice-disruptive variants; predicts impact of engineered mutations on splicing efficiency [62]. |
| Motif-Oriented Tools | Not specified in results | Evaluates mutations affecting splicing regulatory elements (ESEs, ESSs, ISEs, ISSs) [62]. |
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagent Solutions for Splicing and Logic Gate Analysis
| Reagent / Material | Function | Application Context |
|---|---|---|
| Minigene Reporter Plasmid | A plasmid-encoded 2-intron/3-exon construct to monitor splicing efficiency of a specific exon [61]. | Validating ribozyme activity and assessing the impact of mutations on splicing. |
| Modified U1 snRNA Plasmid | Expresses U1 snRNA with compensatory mutations to improve recognition of mutant 5' splice-sites [61]. | Therapeutic suppression of mutation-induced splicing defects; a tool to modulate input signals. |
| Split Intein System | Pairs of intein fragments fused to split protein domains; splicing reconstitutes functional protein [64] [63]. | Core component for constructing AND gates in protein-based biocomputing. |
| TALE Activators | Transcriptional activator-like effectors (TALEs) designed to bind specific DNA sequences [63]. | Activating transcription from synthetic promoters in genetic circuits. |
| Orthogonal TALE | A TALE computationally designed to have minimal cross-reactivity with the host genome (e.g., TAL118) [63]. | Reduces off-target effects in genetic circuits, improving output signal fidelity. |
Experimental Protocols
Protocol 1: Minigene Splicing Assay for Ribozyme Efficiency
This protocol adapts a cellular reporter assay to monitor the splicing efficiency of engineered trans-splicing ribozymes [61].
Reagents and Buffers
- DMEM Culture Medium: Supplemented with 10% FBS, 50 U/mL penicillin, and 50 µg/mL streptomycin [61].
- Transfection Reagent: Compatible with the cell line used (e.g., HeLa, HEK293).
- RNAase-free Water: Treated with DEPC and autoclaved.
- 2x Formamide Loading Dye: 95% formamide, 18 mM EDTA, 0.02% bromophenol blue, 0.02% xylene cyanol.
- Urea-PAGE Solution: 10% 19:1 bis/acrylamide, 7 M urea, 1x TBE.
Procedure
Reporter and Ribozyme Co-transfection:
- Seed HeLa or HEK293 cells in a 12-well plate at a density of (2.5 \times 10^5) cells/mL and incubate overnight [61].
- For each sample, prepare Solution I: Mix 0.2 µg of minigene reporter plasmid, 1.8 µg of ribozyme expression plasmid (or empty vector control), and 100 µL of transfection medium.
- Prepare Solution II: A master mix of 100 µL transfection medium and 4 µL transfection reagent per sample.
- Combine Solutions I and II, vortex for 15 seconds, and incubate at room temperature for 5 minutes.
- Add the 200 µL transfection mix to each well containing cells and fresh medium.
RNA Isolation and Analysis:
- 48 hours post-transfection, isolate total cellular RNA using a standard method (e.g., TRIzol).
- Perform Reverse Transcription using a gene-specific primer or oligo-dT primer to generate cDNA.
- Analyze splicing efficiency via semi-quantitative RT-PCR or primer extension using primers flanking the splice junction [61].
- Resolve PCR products on a 2% agarose gel. For higher resolution, use urea-PAGE (8%) for primer extension products [61].
Data Interpretation:
- Quantify band intensities corresponding to spliced and unspliced products using densitometry.
- Calculate splicing efficiency as: (\frac{\text{[Spliced Product]}}{\text{[Spliced Product] + [Unspliced Product]}} \times 100\%).
Protocol 2: Characterizing a Split-Intein-Based AND Gate in Mammalian Cells
This protocol details the implementation and characterization of a two-input AND gate using a split-intein strategy to reconstitute a functional transcriptional activator, as demonstrated with TALE proteins [63].
Reagents and Buffers
- Split Intein-TALE Constructs: N- and C-terminal fragments of a protein (e.g., TALE) fused to appropriate split intein halves [63].
- Reporter Plasmid: Contains a promoter with the corresponding TALE binding site upstream of a fluorescent protein gene (e.g., AmCyan-CFP) [63].
- Flow Cytometry Buffer: Dulbecco’s phosphate-buffered saline containing 0.1% fetal bovine serum.
Procedure
Cell Transfection and Induction:
- Seed mammalian cells (e.g., U-2 OS) and transfect with three plasmids: the N-terminal intein-TALE fragment, the C-terminal intein-TALE fragment, and the fluorescent reporter plasmid [63].
- Include controls for basal activity: transfect each intein-TALE fragment individually with the reporter.
- For inducible systems, apply relevant chemical inducers (e.g., arabinose, AHL) to activate expression of the split fragments [64].
Flow Cytometry Analysis:
- 48 hours post-transfection, trypsinize cells, pellet them, and resuspend in flow cytometry buffer.
- Analyze approximately 50,000-100,000 live cells using a flow cytometer (e.g., LSRII) [63].
- Gate for cells successfully transfected based on a constitutive marker (e.g., mCherry from a co-transfected plasmid).
- Within the mCherry-positive population, determine the percentage of cells expressing the output fluorescent protein (e.g., CFP) and the mean fluorescence intensity.
Logic Gate Performance Calculation:
- Dynamic Range: Calculate as the ratio of the mean CFP fluorescence in the presence of both inputs to the mean CFP fluorescence in the presence of only one input.
- Leakiness: Defined as the percentage of CFP-positive cells (or mean fluorescence) in single-input control conditions.
- Response Time: Perform a time-course experiment after induction, measuring fluorescence output at multiple time points to determine the delay until a stable ON state is reached.
Workflow and Pathway Diagrams
Minigene Splicing Assay Workflow
The diagram below illustrates the key steps in the minigene splicing assay protocol for evaluating ribozyme efficiency.
Split-Intein AND Gate Mechanism
The diagram below illustrates the molecular mechanism of a split-intein-based AND gate for biological computation.
The precise measurement of splicing efficiency and logic gate performance is fundamental to the advancement of biocomputing systems based on trans-splicing group I introns. The application notes and detailed protocols provided here offer researchers a standardized framework for characterizing these critical functional readouts. By employing robust quantitative assays, such as the minigene splicing reporter and flow-cytometry-based gate characterization, scientists can iteratively design, optimize, and validate increasingly complex and reliable genetic circuits. The integration of these functional assessments will accelerate the development of sophisticated biological computers for therapeutic and diagnostic applications.
The advent of programmable gene editing tools, particularly CRISPR-Cas systems, has revolutionized therapeutic development by enabling precise modification of genetic sequences [66]. However, the clinical translation of these technologies is significantly hampered by concerns about off-target genotoxicity, where unintended modifications occur at sites other than the intended target [66] [67]. Similarly, in the emerging field of trans-splicing group I introns for biocomputing and therapeutic applications, understanding and controlling off-target effects is paramount for ensuring specificity and safety [7] [68]. While CRISPR-Cas systems operate at the DNA level, trans-splicing group I introns function at the RNA level, yet both face the fundamental challenge of achieving high specificity in the complex cellular environment.
Off-target activity spans a spectrum of consequences, from point mutations to large-scale chromosomal rearrangements [69]. In therapeutic contexts, even low-frequency off-target events can be detrimental if they affect critical genomic regions such as tumor suppressor genes or proto-oncogenes [69]. Regulatory agencies including the FDA and EMA now require comprehensive assessment of both on-target and off-target effects as a prerequisite for clinical approval of gene editing therapies [69] [70]. This application note provides a structured framework for assessing off-target effects, with specific protocols and analytical tools applicable to both DNA-targeting CRISPR systems and RNA-targeting trans-splicing ribozymes.
Quantitative Characterization of Group I Intron Systems
Table 1: Comparative Analysis of Group I Intron Ribozymes for Trans-Splicing Applications
| Ribozyme Source | Subgroup | Size (nt) | Natural Context | Optimal Trans-Design | In Vitro Efficiency | In Vivo Efficiency |
|---|---|---|---|---|---|---|
| Azoarcus BH72 | IC3 | 205 | tRNAIle anticodon stem | Resembles natural cis-splicing context with base-pairing between substrate 5′ portion and ribozyme 3′ exon | High under near-physiological conditions | Low in E. coli |
| Tetrahymena thermophila | IC1 | ~400 | 16S rRNA | Classical design with Extended Guide Sequence (EGS) | High | High in multiple systems |
| Fusarium oxysporum | ID1 | 1237 | cob transcript | Classical Tetrahymena design with EGS | ~70% of pre-RNA spliced after 1 hour | Not tested |
The group I intron from Azoarcus represents a particularly attractive platform for biocomputing applications due to its compact size (205 nt) and rapid folding kinetics compared to the larger Tetrahymena ribozyme [7]. Under near-physiological in vitro conditions, the Azoarcus ribozyme achieves trans-splicing efficiencies comparable to the Tetrahymena ribozyme when both are designed with their preferred secondary structure interactions [7]. Notably, the optimal design for the Azoarcus ribozyme differs from the established Tetrahymena design, emphasizing the importance of ribozyme-specific optimization [7].
Recent work has demonstrated the adaptation of group I introns from pathogenic fungi for trans-splicing applications. The Fusarium oxysporum group I intron, located in the cytochrome b (cob) transcript, exhibits robust self-splicing activity in vitro, with approximately 70% of pre-RNA spliced after 60 minutes [68]. This ribozyme has been successfully converted to a trans-splicing format using the classical design principle originally developed for the Tetrahymena ribozyme, including an extended guide sequence (EGS) to optimize ribozyme-substrate hybridization [68].
Experimental Protocols for Off-Target Assessment
Protocol 1: In Vitro Splice Site Selection Assay for Trans-Splicing Ribozymes
Purpose: To identify preferred splice sites and potential off-target sites for trans-splicing group I intron ribozymes on a model substrate mRNA.
Materials:
- Trans-splicing ribozyme with randomized substrate recognition sequence
- Chloramphenicol acetyl transferase (CAT) mRNA or other model substrate
- Reverse transcriptase and PCR reagents
- NGS library preparation kit
Procedure:
- Incubate the ribozyme pool (with randomized recognition sequence) with the target substrate RNA under near-physiological conditions (e.g., 100 mM NaCl, 5-10 mM MgCl₂, pH 7.5, 37°C) for 30-60 minutes [7].
- Extract and purify RNA after the reaction.
- Perform reverse transcription with a primer specific to the ribozyme's 3′ exon.
- Amplify trans-splicing products using PCR with primers complementary to the ribozyme's 3′ exon and the substrate RNA [7].
- Prepare NGS libraries and sequence to identify splice sites.
- Analyze sequence data to determine frequency and distribution of splice sites.
Analysis: The resulting sequences will reveal preferred splice sites based on abundance in the dataset. Sites with significant representation indicate either on-target or potential off-target activity, depending on the intended application.
Protocol 2: Fluorescence-Based Reporter Assay for Trans-Splicing Efficiency
Purpose: To quantitatively measure trans-splicing efficiency and screen for potential inhibitors or enhancers in a high-throughput format.
Materials:
- Fluorescent reporter plasmid with target sequence
- Trans-splicing ribozyme construct
- Appropriate cell line or in vitro transcription system
- Fluorescence plate reader
- RT-qPCR reagents for validation
Procedure:
- Design a fluorescent reporter system where successful trans-splicing activates or changes fluorescence output [68].
- For in vitro applications: transcribe both substrate and ribozyme RNAs, incubate together, and measure fluorescence development over time.
- For cellular applications: co-transfect reporter construct and ribozyme expression vector into appropriate cells.
- Monitor fluorescence intensity at regular intervals (e.g., 24, 48, 72 hours) using a plate reader.
- Normalize fluorescence readings to control conditions (e.g., no ribozyme, inactive ribozyme mutant).
- Confirm splicing efficiency using RT-qPCR on a subset of samples [68].
Analysis: Calculate trans-splicing efficiency as the fold-increase in fluorescence compared to negative controls. This assay can be adapted to high-throughput screening of multiple ribozyme designs or small molecule modulators.
Protocol 3: CIRCLE-Seq for Comprehensive Off-Target Profiling
Purpose: To identify potential off-target sites of CRISPR-Cas systems through in vitro cleavage and sequencing.
Materials:
- Genomic DNA from target cells
- Cas9-gRNA RNP complex
- CIRCLE-Seq library preparation kit
- Next-generation sequencing platform
Procedure:
- Extract high-molecular-weight genomic DNA from relevant cell types.
- Incubate genomic DNA with pre-formed Cas9-gRNA ribonucleoprotein (RNP) complexes.
- Process cleaved DNA fragments through the CIRCLE-Seq workflow, which involves circularization of non-cleaved DNA, digestion of linear fragments, and amplification of cleaved sites [70].
- Sequence resulting libraries using NGS.
- Analyze sequencing data to map cleavage sites across the genome.
Analysis: Identify off-target sites with significant read accumulation compared to negative controls (e.g., no Cas9, catalytically dead Cas9). Validate top candidate sites using amplicon sequencing in actual edited cells.
Computational Prediction and Analysis Tools
Table 2: Computational Tools for Off-Target Analysis in Genome Editing
| Tool Name | Application | Methodology | Advantages | Limitations |
|---|---|---|---|---|
| CRISPOR | gRNA design and off-target prediction | Genome-wide scanning for sequences with similarity to target | User-friendly interface, integrates multiple scoring algorithms | Limited to CRISPR systems |
| CAST-Seq | Detection of chromosomal rearrangements | Amplification and sequencing of junction fragments between different genomic loci | Specifically designed to identify large structural variations | May miss deletions not involving known off-target sites |
| GUIDE-seq | Unbiased off-target discovery | Integration of oligonucleotide tags into double-strand breaks | Genome-wide, unbiased mapping of nuclease activity | Requires delivery of double-stranded oligodeoxynucleotides |
| ICE (Inference of CRISPR Edits) | Analysis of editing efficiency | Decomposition of Sanger sequencing chromatograms | Fast, cost-effective for candidate site validation | Limited to predefined target sites |
| LAM-HTGTS | Translocation detection | Linear amplification-mediated high-throughput genome-wide translocation sequencing | Sensitive detection of chromosomal translocations | Complex workflow |
Current evidence suggests that no single computational tool can accurately predict all off-target events, particularly low-frequency editing [67]. Therefore, a combination of in silico prediction and experimental validation is recommended for comprehensive off-target assessment. For trans-splicing ribozymes, computational prediction involves identifying potential off-target RNAs with complementarity to the ribozyme's internal guide sequence (IGS), particularly at sites containing the essential U-G base pair required for splicing [7].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Off-Target Assessment Studies
| Reagent Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| Nucleases | SpCas9, HiFi Cas9, Cas12 | Target DNA cleavage | High-fidelity variants reduce off-target but may lower on-target efficiency |
| Group I Intron Ribozymes | Azoarcus, Tetrahymena, Fusarium oxysporum | RNA trans-splicing | Species-specific optimization required for efficient trans-splicing |
| gRNA Modifications | 2'-O-methyl analogs (2'-O-Me), 3' phosphorothioate bonds (PS) | Enhance stability and reduce off-target effects | Chemical modifications improve specificity and editing efficiency |
| Detection Reagents | GUIDE-seq dsODN, CIRCLE-seq adapter oligos | Tagging and capturing editing events | Essential for unbiased genome-wide off-target discovery |
| Reporter Systems | Fluorescent splicing reporters, SURVEYOR nuclease, T7E1 | Detect editing efficiency | Fluorescent systems enable real-time monitoring and HTS |
| Sequencing Kits | Amplicon sequencing kits, WGS libraries | Characterize editing outcomes | Amplicon-seq targets specific sites; WGS provides comprehensive coverage |
Safety Assessment and Risk Mitigation Strategies
Diagram 1: Comprehensive Safety Assessment Workflow for Therapeutic Genome Editing
Effective risk mitigation begins with careful gRNA or IGS design. For CRISPR systems, this involves selecting guides with high specificity scores, minimal off-target potential, and optimal GC content (40-60%) [70]. For trans-splicing ribozymes, the internal guide sequence should be designed to maximize complementarity to the intended target while minimizing similarity to non-target transcripts [7]. Extended guide sequences (EGS) can enhance specificity but must be optimized for each ribozyme type [7] [68].
Delivery method and duration of expression significantly impact off-target effects. Transient delivery methods (e.g., RNA or RNP delivery) reduce the window for off-target activity compared to stable plasmid expression [70]. For therapeutic applications, the use of high-fidelity Cas variants such as HiFi Cas9 can substantially reduce off-target editing while maintaining on-target efficiency [69] [70]. Similarly, for trans-splicing applications, ribozyme engineering to enhance specificity, potentially through structure-guided design, represents a promising approach.
Recent studies have revealed that beyond simple indels, CRISPR editing can generate large structural variations (SVs) including kilobase- to megabase-scale deletions and chromosomal rearrangements [69]. These SVs pose significant safety concerns and are often undetected by conventional amplicon sequencing. Methods such as CAST-Seq and LAM-HTGTS have been developed specifically to identify these larger aberrations [69]. Assessment of these genomic alterations should be incorporated into safety evaluation pipelines, particularly for therapeutic development.
Regulatory Considerations and Future Directions
Regulatory agencies increasingly require comprehensive off-target assessment for gene editing therapies. The FDA's review of Casgevy (exa-cel) focused extensively on potential off-target effects, highlighting that individuals with rare genetic variants may be at higher risk [70]. Similarly, therapies based on trans-splicing ribozymes will require thorough characterization of specificity and potential off-target RNA modification.
Future directions for improving specificity include the development of more sophisticated computational prediction algorithms trained on expanded datasets of true off-target sites [67]. For both CRISPR and trans-splicing systems, continued engineering of more specific variants through directed evolution or structure-based design will enhance the therapeutic potential of these technologies. Additionally, standardized reference materials and benchmarking datasets will enable more consistent off-target assessment across studies and platforms [67].
The integration of multiple assessment methods—combining in silico prediction, in vitro profiling, and cell-based validation—provides the most comprehensive approach to evaluating off-target effects. This multi-layered strategy is essential for advancing both DNA-targeting CRISPR therapies and RNA-targeting trans-splicing systems toward safe clinical application.
Conclusion
Trans-splicing group I introns have evolved from curious genetic elements into versatile and programmable platforms for synthetic biology. Their unique ability to be engineered for specific RNA recognition and sequence replacement makes them ideal for applications ranging from complex cellular logic computation to the precise repair of disease-causing mutations. Key advancements in understanding their structure, optimizing their efficiency with tools like Extended Guide Sequences, and validating their function in therapeutic contexts have solidified their potential. Future directions will likely focus on improving in vivo delivery and stability, expanding the library of orthogonal ribozymes for more complex circuits, and moving promising therapeutic candidates, like those for NF1, closer to clinical reality. The integration of machine learning for design and the exploration of novel introns from diverse species will further unlock the potential of these RNA machines, paving the way for a new era of RNA-based diagnostics, therapeutics, and biocomputing.
References
- 1. Group I catalytic intron [https://en.wikipedia.org/wiki/Group_I_catalytic_intron]
- 2. Group I introns: Structure, splicing and their applications in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10964413/]
- 3. Nuclear group I introns in self-splicing and beyond - Mobile DNA [https://mobilednajournal.biomedcentral.com/articles/10.1186/1759-8753-4-17]
- 4. The natural history of group I introns [https://www.sciencedirect.com/science/article/abs/pii/S016895250400335X]
- 5. Bacterial group I introns: mobile RNA catalysts [https://mobilednajournal.biomedcentral.com/articles/10.1186/1759-8753-5-8]
- 6. Phylogenetic analyses suggest reverse splicing spread of ... [https://bmcecolevol.biomedcentral.com/articles/10.1186/1471-2148-5-68]
- 7. Trans-splicing with the group I intron ribozyme from Azoarcus [https://pmc.ncbi.nlm.nih.gov/articles/PMC3895272/]
- 8. Repair of Mutated NF1 mRNA with Trans-Splicing Group I ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12427287/]
- 9. Self-splicing RNA circularization facilitated by intact group I ... [https://www.nature.com/articles/s41467-025-62607-y]
- 10. RNA splicing [https://en.wikipedia.org/wiki/RNA_splicing]
- 11. Structural Divergence of the Group I Intron Binding Surface ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4882457/]
- 12. Homing endonuclease [https://en.wikipedia.org/wiki/Homing_endonuclease]
- 13. Homing endonucleases: from basics to therapeutic applications [https://pmc.ncbi.nlm.nih.gov/articles/PMC11115532/]
- 14. Structural Basis for the Propagation of Homing ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2022.855511/full]
- 15. Accurate, comprehensive database of group I introns and ... [https://pubmed.ncbi.nlm.nih.gov/39968379/]
- 16. Lineage barcoding in mouse with homing CRISPR - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8049957/]
- 17. Therapeutic Applications of Group I Intron-Based Trans- ... [https://www.advancedsciencenews.com/therapeutic-applications-group-intron-based-trans-splicing-ribozymes/]
- 18. Evolutionary Insights into RNA trans-Splicing in Vertebrates [https://pmc.ncbi.nlm.nih.gov/articles/PMC4824033/]
- 19. Therapeutic applications of trans-splicing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7737522/]
- 20. Origin and evolution of spliceosomal introns - Biology Direct [https://biologydirect.biomedcentral.com/articles/10.1186/1745-6150-7-11]
- 21. Characterization of trans-spliced chimeric RNAs [https://pmc.ncbi.nlm.nih.gov/articles/PMC11155486/]
- 22. Volume 21 Issue 5, May 2025 [https://www.nature.com/nchembio/volumes/21/issues/5]
- 23. Exploring the space of self-reproducing ribozymes using ... [https://www.nature.com/articles/s41467-025-63151-5]
- 24. Ribozyme Assays to Quantify the Capping Efficiency of In ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8879150/]
- 25. Programming RNA splicing for complex cellular logic ... [https://communities.springernature.com/posts/programming-rna-splicing-for-complex-cellular-logic-computation]
- 26. Programmable trans-splicing riboregulators for complex ... [https://pubmed.ncbi.nlm.nih.gov/39747656/]
- 27. Repair of Mutated NF1 mRNA with Trans-Splicing Group I ... [https://pubmed.ncbi.nlm.nih.gov/40940846/]
- 28. Gene Therapy for Neurofibromatosis: A New Frontier in ... [https://ventures.jhu.edu/news/gene-therapy-for-neurofibromatosis-a-new-frontier-in-treatment/]
- 29. Johns Hopkins Team Creates Targeted Gene Therapy ... [https://www.hopkinsmedicine.org/news/articles/2025/10/johns-hopkins-team-creates-targeted-gene-therapy-vector-for-neurofibromatosis-type-1-tumors]
- 30. Advances in RNA-based therapeutics: current breakthroughs ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12592931/]
- 31. Design and Experimental Evolution of trans-Splicing Group ... [https://www.mdpi.com/1420-3049/22/1/75]
- 32. Reevaluating therapeutic space for small catalytic RNAs [https://www.sciencedirect.com/science/article/pii/S2162253124003184]
- 33. Characterization of group I introns in generating circular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11879131/]
- 34. CellREADR: An ADAR-based RNA sensor-actuator device [https://www.sciencedirect.com/science/article/abs/pii/S0076687924006128]
- 35. Engineering modular intracellular protein sensor-actuator ... [https://www.nature.com/articles/s41467-018-03984-5]
- 36. Group I Intron as a Potential Target for Antifungal ... [https://www.mdpi.com/1420-3049/28/11/4460]
- 37. Computational prediction of efficient splice sites for trans ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3285945/]
- 38. External guide sequence technology: a path to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4600001/]
- 39. Principles for the design of multicellular engineered living ... [https://pubmed.ncbi.nlm.nih.gov/35274072/]
- 40. Robust prediction of synthetic gRNA activity and cryptic ... [https://www.nature.com/articles/s41467-025-59947-0]
- 41. Principles of in vitro selection of ribozymes from random ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11999736/]
- 42. A split ribozyme that links detection of a native RNA to ... [https://www.nature.com/articles/s41467-023-36073-3]
- 43. Orthogonal intercellular signaling for programmed spatial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4731010/]
- 44. Protein Folding in the Cell: Challenges and Progress - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3072030/]
- 45. A Method for Direct Measurement of Protein Stability In Vivo [https://pmc.ncbi.nlm.nih.gov/articles/PMC2892803/]
- 46. Intracellular delivery [https://en.wikipedia.org/wiki/Intracellular_delivery]
- 47. Physical Methods for Intracellular Delivery [https://pmc.ncbi.nlm.nih.gov/articles/PMC4449156/]
- 48. Design and Experimental Evolution of trans-Splicing Group I ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6155759/]
- 49. Combinatorial optimization of mRNA structure, stability ... [https://www.nature.com/articles/s41467-022-28776-w]
- 50. Comprehensive datasets for RNA design, machine learning ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12215848/]
- 51. Snapshots of the second-step self-splicing of Tetrahymena ... [https://www.nature.com/articles/s41467-023-36724-5]
- 52. Modular engineering of a Group I intron ribozyme - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC137077/]
- 53. sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology... [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/azoarcus]
- 54. Increased Ribozyme Activity in Crowded Solutions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3908428/]
- 55. A Quantitative High-Throughput In Vitro Splicing Assay ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3302438/]
- 56. Unexpected intron plasticity and trans-splicing capability ... [https://www.sciencedirect.com/science/article/pii/S0020751925001572]
- 57. Integrative transcriptome sequencing identifies trans ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3875859/]
- 58. Co-transcriptional gene regulation in eukaryotes and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11199108/]
- 59. mRNA medicine: Recent progresses in chemical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12311907/]
- 60. mRNA therapeutics: Transforming medicine through ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12590236/]
- 61. A reporter based cellular assay for monitoring splicing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8643280/]
- 62. Genome-wide functional annotation and interpretation of ... [https://www.nature.com/articles/s10038-025-01424-z]
- 63. Two- and three-input TALE-based AND logic computation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3834826/]
- 64. A systematic approach to inserting split inteins for Boolean ... [https://www.nature.com/articles/s41467-021-22404-9]
- 65. Gate Performance Characteristics and Parameters | PDF [https://www.scribd.com/document/657846376/Gate-performance-characteristics-and-parameters]
- 66. Off-target effects in CRISPR-Cas genome editing for ... [https://www.sciencedirect.com/science/article/pii/S2162253125001908]
- 67. Tools for experimental and computational analyses of off- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8049448/]
- 68. as a Potential Target for Antifungal Compounds... Group I Intron [https://pmc.ncbi.nlm.nih.gov/articles/PMC10254303/]
- 69. The hidden risks of CRISPR/Cas: structural variations and ... [https://www.nature.com/articles/s41467-025-62606-z]
- 70. CRISPR Off-Target Editing: Prediction, Analysis, and More [https://www.synthego.com/blog/crispr-off-target-editing/]