CGE-LIF: The Ultimate Guide to High-Sensitivity Biomolecular Analysis for Biopharmaceutical R&D
This comprehensive guide provides researchers and drug development professionals with an in-depth exploration of Capillary Gel Electrophoresis with Laser-Induced Fluorescence detection (CGE-LIF).
CGE-LIF: The Ultimate Guide to High-Sensitivity Biomolecular Analysis for Biopharmaceutical R&D
Abstract
This comprehensive guide provides researchers and drug development professionals with an in-depth exploration of Capillary Gel Electrophoresis with Laser-Induced Fluorescence detection (CGE-LIF). It covers the fundamental principles and components of the technology, details step-by-step methodologies and key applications in biopharmaceutical analysis, presents practical troubleshooting and optimization strategies, and offers a critical comparison with alternative techniques. The article serves as a current, authoritative resource for implementing and validating CGE-LIF to characterize critical quality attributes of proteins, oligonucleotides, and gene therapies.
What is CGE-LIF? Principles, Components, and Core Advantages Explained
Capillary Gel Electrophoresis (CGE) is a high-resolution analytical technique that separates charged molecules, primarily biopolymers like DNA, RNA, proteins, and oligonucleotides, based on their size-to-charge ratio within a capillary filled with a viscous sieving matrix. This article, framed within a broader thesis on Capillary Gel Electrophoresis with Laser-Induced Fluorescence detection (CGE-LIF), details the critical role of the sieving matrix in achieving size-based separation, its applications in drug development, and provides detailed protocols for researchers.
The Sieving Matrix: Principles and Function
The sieving matrix is a critical component in CGE, replacing the traditional cross-linked polyacrylamide gel used in slab-gel electrophoresis with a dynamic, polymer-based solution. This matrix creates a porous network through which analytes migrate under an applied electric field. Separation is achieved because smaller molecules navigate the pores more readily than larger ones, effectively sieving molecules by hydrodynamic size.
Key Functions:
- Size-Based Separation: Creates a size-dependent mobility regime.
- Suppresses Electroosmotic Flow (EOF): Many polymer matrices coat the capillary wall, reducing EOF and improving separation reproducibility.
- Minimizes Analyte Adsorption: Coats the capillary surface to prevent sample adsorption.
- Compatibility with On-column Detection: Allows for real-time, on-capillary detection methods like LIF.
Quantitative Comparison of Common Sieving Matrices
The choice of matrix depends on the analyte, required resolution, and detection method. The table below summarizes key characteristics.
Table 1: Properties of Common Sieving Matrices for CGE
| Matrix Polymer | Typical Concentration Range | Effective Separation Range (dsDNA, bp) | Key Advantages | Primary Applications |
|---|---|---|---|---|
| Linear Polyacrylamide (LPA) | 2-6% (w/v) | 10 - 1,000+ | High resolution, low viscosity, excellent UV transparency | DNA fragment analysis, Sanger sequencing, protein analysis. |
| Polyethylene Oxide (PEO) | 0.5-2% (w/v) | 20 - 10,000+ | Low viscosity, good for large DNA fragments, suppresses EOF effectively. | Analysis of PCR products, large DNA fragments. |
| Cellulose Derivatives (e.g., HPMC) | 0.1-1% (w/v) | 100 - 10,000 | Low cost, good for routine size analysis, moderate EOF suppression. | Routine QC of DNA samples, plasmid analysis. |
| Pullulan | 2-4% (w/v) | 10 - 1,000 | Excellent resolution for small fragments, stable performance. | Oligonucleotide analysis, small DNA/RNA fragments. |
| Polyvinylpyrrolidone (PVP) | 1-3% (w/v) | 50 - 5,000 | Good dynamic coating, compatible with various buffers. | General-purpose DNA and protein separations. |
Application Notes in Drug Development and Biopharma
CGE-LIF is indispensable in biopharmaceutical development due to its high sensitivity, quantitative capabilities, and automation.
- Oligonucleotide Therapeutics Purity and Impurity Analysis: CGE-LIF is the gold standard for assessing the purity of synthetic oligonucleotides (e.g., siRNA, ASOs) by separating the full-length product from shorter failure sequences (N-1, N-2) and other impurities with single-nucleotide resolution.
- Gene Therapy Vector Analysis (AAV Capsid Proteins): CGE-LIF enables the identity and purity testing of Adeno-Associated Virus (AAV) capsid proteins (VP1, VP2, VP3). The sieving matrix separates these proteins based on size, providing a critical quality attribute for vector potency and safety.
- Biologic Charge Variant Analysis (when coupled with cIEF): While primarily for size, CGE principles apply to capillary isoelectric focusing (cIEF) for monitoring charge heterogeneity of monoclonal antibodies and other proteins.
- PCR Product Quality Control: Rapid, high-throughput sizing and quantitation of PCR amplicons for genetic testing and bioprocess monitoring.
Detailed Experimental Protocols
Protocol 1: CGE-LIF for Oligonucleotide Purity Analysis
Objective: To separate and quantify a 25-mer synthetic oligonucleotide from its N-1 and N-2 failure sequences.
The Scientist's Toolkit: Table 2: Essential Reagents and Materials
| Item | Function | Example/Note |
|---|---|---|
| CGE-LIF Instrument | Performs electrophoresis, separation, and detection. | System with LIF detector (e.g., λex 488 nm / λem 520 nm). |
| Fused-Silica Capillary | Separation channel. | 50 µm ID, 30-50 cm total length (20-40 cm to detector). |
| Linear Polyacrylamide (LPA) Matrix | Sieving matrix for high-resolution separation. | 4% (w/v) LPA in 1x TBE with 7M Urea. |
| Urea | Denaturant. Prevents secondary structure in oligonucleotides. | Ensures separation is based solely on length. |
| TBE Buffer (Tris-Borate-EDTA) | Running buffer. Provides conductivity and maintains pH. | Typically 1x concentration (89 mM Tris, 89 mM Boric Acid, 2 mM EDTA, pH 8.3). |
| Fluorescent Intercalating Dye | Binds nucleic acids for LIF detection. | SYBR Green I, GelGreen, or proprietary dyes. |
| Internal Size Standard | Allows for precise fragment sizing. | Fluorescently labeled DNA ladder covering 10-100 bp. |
| Formamide or EDTA Solution | Sample diluent/stop solution. | Contains a co-fluorescent marker for injection tracking. |
Methodology:
- Capillary Conditioning: Flush new capillary sequentially with 1 M HCl (10 min), deionized water (5 min), 1 M NaOH (20 min), water (5 min), and separation matrix (10 min). Between runs, flush with matrix for 3 min.
- Matrix Preparation: Prepare 4% LPA sieving matrix in 1x TBE buffer containing 7M Urea and the recommended concentration of intercalating dye (e.g., 1:10,000 dilution of SYBR Green I stock). Filter through a 0.45 µm syringe filter. Degas briefly.
- Sample Preparation: Dilute the oligonucleotide sample to ~0.1 µg/µL in deionized water or a formamide/EDTA solution containing a co-injection marker. Mix the internal size standard separately.
- Instrument Parameters:
- Injection: Electrokinetic injection: 5 kV for 10 seconds.
- Separation Voltage: -15 kV (reverse polarity for negatively charged DNA).
- Temperature: 30°C.
- Detection: LIF, λex 488 nm / λem 520 nm.
- Run: Place inlet vial in sample, outlet vial in buffer. Start the run. The co-injection marker provides a reference peak. The internal standard is run in a separate sequence for calibration.
- Data Analysis: Integrate peaks. Calculate the percentage of main peak (25-mer) relative to the total integrated area of all oligonucleotide peaks. Use the size standard curve to confirm the size of each peak.
Protocol 2: CGE-LIF for AAV Capsid Protein Ratio Analysis
Objective: To separate and determine the VP1:VP2:VP3 ratio of denatured AAV capsid proteins.
Methodology:
- Sample Denaturation: Incubate purified AAV sample (~1e12 vg/µL) with denaturation buffer (e.g., containing SDS and β-mercaptoethanol) at 95°C for 5 minutes.
- Capillary and Matrix: Use a pre-coated capillary (e.g., SDS-MW coating). Fill with a replaceable SDS-based gel matrix (e.g., 1% PVP or dextran in Tris-Tricine-SDS buffer).
- Sample Preparation: Dilute denatured sample 1:10 in running buffer.
- Instrument Parameters:
- Injection: Pressure injection: 0.5 psi for 20 seconds.
- Separation Voltage: +15 kV.
- Temperature: 25°C.
- Detection: LIF with post-column labeling or native fluorescence if applicable.
- Run and Analysis: Separate proteins by size (VP3 ~60 kDa, VP2 ~72 kDa, VP1 ~87 kDa). Integrate peak areas to calculate the percentage of each VP protein, a critical quality attribute.
Visualization of CGE-LIF Workflow and Separation Mechanism
Within the framework of Capillary Gel Electrophoresis with Laser-Induced Fluorescence detection (CGE-LIF) research, achieving zeptomole (10⁻²¹ mol) sensitivity represents a frontier in trace analysis. This level of detection is critical for applications such as quantifying low-abundance biomarkers, analyzing single cells, and ensuring the purity of biopharmaceuticals like oligonucleotide therapeutics and gene therapies. LIF's unparalleled sensitivity stems from its ability to excite fluorophores with a high-intensity laser source and collect emitted photons with minimal background noise, a principle leveraged to its extreme in optimized CGE-LIF systems.
Key Principles and Advances
Modern CGE-LIF systems achieve zeptomole sensitivity through a multi-faceted approach:
- High-Power Lasers: Utilization of solid-state lasers (e.g., 488 nm, 520 nm) providing stable, focused excitation.
- Low-Background Capillaries: Coated capillaries (e.g., linear polyacrylamide) reduce analyte adsorption and scatter.
- High-Efficiency Fluorophores: Use of cyanine dyes (Cy5, Alexa Fluor series) with high quantum yields and photostability.
- Advanced Detection Optics: Confocal microscope setups with high numerical aperture objectives and spectral filters minimize stray light.
- Low-Noise Detectors: Photon-counting photomultiplier tubes (PMTs) or avalanche photodiodes (APDs) are essential.
Table 1: Quantitative Performance Metrics of Advanced CGE-LIF Systems
| Parameter | Typical Performance Range | Notes |
|---|---|---|
| Limit of Detection (LOD) | 1 - 100 zeptomoles | Depends on fluorophore and background. |
| Linear Dynamic Range | 3 - 5 orders of magnitude | From low zmol to high fmol. |
| Separation Efficiency | > 1 million theoretical plates | For dsDNA fragments in gel-filled capillaries. |
| Run-to-Run Precision (RSD) | < 2% (migration time) | Critical for quantitative analysis. |
| Laser Power Stability | < 1% fluctuation over 1 hr | Essential for baseline stability. |
Application Notes
Note 1: Analysis of Trace-Level Impurities in Oligonucleotide Therapeutics
Challenge: Detecting and quantifying impurity species (e.g., N-1, N+1 failure sequences) at levels <0.1% in a bulk synthesized oligonucleotide sample. CGE-LIF Solution: A highly sensitive, size-based separation using a gel polymer matrix (e.g., POP-7) with a fluorescent intercalating dye (e.g., SYBR Gold) or end-labeled primers. Outcome: Zeptomole sensitivity enables the detection of single-molecule events in highly diluted samples, providing a purity profile far exceeding UV absorbance detection.
Note 2: Single-Cell Genomic and Proteomic Analysis
Challenge: Quantifying nucleic acids or proteins from a single cell, where total amounts can be in the attomole to zeptomole range. CGE-LIF Solution: After single-cell lysis and pre-column fluorescent labeling (e.g., with a FAM NHS ester for proteins), CGE-LIF separates and quantifies molecules like microRNAs or post-translationally modified proteins. Outcome: Enables the study of cellular heterogeneity in cancer research and neurobiology without the need for bulk population analysis.
Experimental Protocols
Protocol 1: CGE-LIF for DNA Fragment Analysis at Zeptomole Sensitivity
Objective: To separate and detect a DNA ladder (10-1000 bp) with LOD in the zeptomole range. Materials: See "The Scientist's Toolkit" below.
Methodology:
- Capillary Preparation: Install a 50 µm ID, 30 cm effective length (40 cm total) capillary coated for gel electrophoresis. Fill with a sieving polymer matrix (e.g., a commercial replaceable gel) using a high-pressure syringe.
- Sample Preparation: Dilute a fluorescently end-labeled (e.g., 6-FAM) DNA ladder to a series of concentrations from 1 fM to 10 aM in deionized water or a compatible low-ionic-strength buffer.
- Instrument Setup: Configure the LIF detector. Set laser excitation to 488 nm, emission filter to 520 nm (± 10 nm). Set PMT voltage to its optimal, high-gain setting. Set data collection rate to 10 Hz.
- Injection: Hydrodynamically inject sample at 3.5 kPa for 10 seconds. This typically introduces a few picoliters, corresponding to zeptomole amounts.
- Electrophoresis: Apply a separation voltage of 15 kV (reverse polarity) at a constant temperature of 25°C. Run time: ~20 minutes.
- Data Analysis: Use instrument software to plot fluorescence intensity vs. migration time. Construct a calibration curve using peak area from the known concentration series to determine LOD (typically S/N > 3).
Protocol 2: Impurity Profiling of Synthetic siRNA
Objective: To quantify short deletion/insertion impurities in a synthesized siRNA duplex. Materials: siRNA sample, fluorescent intercalating dye (e.g., SYTOX Orange), CGE-LIF system with 520 nm laser, sieving matrix optimized for small RNAs.
Methodology:
- Dye Loading: Prepare a 1 µM stock of SYTOX Orange in DMSO. Mix 5 µL of 10 µM siRNA with 1 µL of dye stock and 94 µL of TE buffer. Incubate in the dark for 15 minutes.
- System Equilibration: Flush capillary with sieving matrix for 3 minutes. Pre-run at 10 kV for 5 minutes to establish a stable baseline.
- Sample Injection: Electrokinetically inject at 5 kV for 10 seconds.
- Separation: Run at 12 kV, 40°C, for 15 minutes.
- Detection & Quantification: Detect impurity peaks migrating just before or after the main siRNA peak. Use peak area relative to the main peak to calculate percent impurity.
Visualization: Workflows and Pathways
Diagram 1: CGE-LIF Core Workflow
Diagram 2: LIF Signal and Noise Control
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function | Example/Note |
|---|---|---|
| Fluorescent Dyes | Covalently tag or intercalate with analyte for detection. | Alexa Fluor 488, Cy5 (covalent); SYBR Gold, SYTOX Orange (intercalating). |
| Sieving Polymer Matrix | Size-based separation medium for nucleic acids or proteins. | Replaceable linear polyacrylamide (LPA), commercial polymers (e.g., POP-7). |
| Coated Capillaries | Minimize electroosmotic flow (EOF) and analyte adsorption. | Capillaries coated with linear polyacrylamide or polyvinyl alcohol (PVA). |
| Capillary Conditioning Kits | Clean and re-condition capillary surface between runs. | Includes sequential solutions of NaOH, HCl, water, and run buffer. |
| Low-Binding Microtubes | Prevent loss of trace analyte via surface adsorption. | Tubes made from polypropylene with polymer additives. |
| High-Purity Run Buffers | Provide consistent ionic strength and pH for separation. | Tris-Borate-EDTA (TBE) or Tris-Acetate-EDTA (TAE), filtered (0.2 µm). |
| Internal Standards | Fluorescently-labeled molecules for migration time and quantification normalization. | Custom-labeled DNA or RNA fragments of known size. |
| Photon-Counting Detector | Converts faint fluorescence emission into an amplified electrical signal. | Photomultiplier Tube (PMT) or Avalanche Photodiode (APD) module. |
Within the context of advancing Capillary Gel Electrophoresis with Laser-Induced Fluorescence detection (CGE-LIF) for biopharmaceutical analysis, the performance and integration of each system component are paramount. This application note details the critical hardware and software elements, from the separation matrix to data interpretation, providing protocols and resources for researchers and drug development professionals engaged in high-sensitivity applications such as monoclonal antibody (mAb) purity assessment, gene therapy vector analysis, and oligonucleotide characterization.
Core System Components & Research Reagent Solutions
The following table summarizes essential materials and their functions for a standard CGE-LIF setup for protein sizing.
Table 1: Key Research Reagent Solutions for CGE-LIF Analysis
| Component/Reagent | Function & Rationale |
|---|---|
| Fused Silica Capillary | The separation channel (typically 20-50 µm ID). Coated capillaries (e.g., polyacrylamide) minimize electroosmotic flow (EOF) and analyte adsorption. |
| Gel Polymer Matrix | A replaceable sieving matrix (e.g., linear polyacrylamide, polyethylene oxide) for size-based separation. Critical for resolution of size variants. |
| Fluorescent Dye (e.g., Pyromine Y, Alexa Fluor 488) | Covalently labels proteins/Nucleic acids pre- or post-separation. Enables LIF detection with high sensitivity (zeptomole levels). |
| LIF Detector (Argon-ion laser, 488 nm) | Excites the fluorophore; emitted light (>510 nm) is collected by a photomultiplier tube (PMT). Provides 100-1000x sensitivity over UV detection. |
| High-Voltage Power Supply (0-30 kV) | Drives the electrophoretic separation. Precise, reversible voltage control is essential for reproducibility and automated runs. |
| Data Acquisition & Analysis Software | Converts analog PMT signal to digital electropherogram, provides tools for peak identification, sizing, and quantitation (e.g., % purity). |
Quantitative Performance Data
Performance metrics for CGE-LIF are system-dependent. The following table generalizes achievable specifications under optimized conditions.
Table 2: Typical CGE-LIF System Performance Metrics
| Parameter | Typical Performance Range | Notes / Conditions |
|---|---|---|
| Detection Limit | 0.1 – 1.0 µg/mL (Proteins) | Using pre-column labeling with a high-quantum-yield fluorophore. |
| Size Resolution (RS) | >1.5 for size variants differing by ~10% | e.g., Separation of 40 kDa and 44 kDa protein fragments. |
| Migration Time RSD | < 1.5% (intra-day) | Dependent on polymer matrix stability and temperature control. |
| Linear Dynamic Range | 2 – 3 orders of magnitude | For quantitation of major and minor variants. |
| Capillary Lifetime | 50 – 200 runs | With proper storage and polymer replacement protocols. |
Detailed Experimental Protocol: mAb Purity and Fragmentation Analysis by CGE-LIF
Protocol 1: Pre-column Labeling and Analysis of a Monoclonal Antibody
Application: Quantification of mAb fragments (non-glycosylated heavy chain, light chain) and aggregates.
I. Materials & Preparation
- Sample: Monoclonal antibody, 1 mg/mL in formulation buffer.
- Labeling Dye: 10 mM solution of amine-reactive fluorescent dye (e.g., Alexa Fluor 488 NHS ester) in DMSO.
- Gel Matrix: 1% (w/v) linear polyacrylamide gel buffer containing 100 mM Tris-Borate, 1% SDS, pH 8.5.
- Running Buffer: Identical to gel matrix buffer.
- Capillary: 50 µm ID, 30 cm total length (20 cm to detector), coated to suppress EOF.
II. Step-by-Step Methodology
- Sample Derivatization:
- Mix 10 µL of mAb sample (1 mg/mL) with 2 µL of 1M sodium bicarbonate buffer (pH 9.0).
- Add 1 µL of 10 mM fluorescent dye solution. Vortex gently.
- Incubate at 25°C in the dark for 30 minutes.
- Quench the reaction by adding 2 µL of 1.5M hydroxylamine (pH 8.5). Incubate for 10 minutes.
Capillary & System Preparation:
- Install a fresh capillary cartridge according to the instrument manual.
- Fill the capillary and anode buffer vial with fresh gel-running buffer.
- Prime the capillary with gel matrix at high pressure (e.g., 50 psi for 3 min).
- Set the instrument temperature to 25°C and the detector to appropriate settings for your fluorophore (e.g., excitation 488 nm, emission 520 nm BP filter).
Electrophoretic Run:
- Dilute the labeled sample 1:10 with deionized water.
- Place the sample in a vial in the autosampler.
- Inject the sample electrokinetically at 5 kV for 10 seconds.
- Apply a separation voltage of 15 kV (reverse polarity, cathode at detector side) for 20 minutes.
- Between runs, rinse the capillary sequentially with 0.1M NaOH (1 min), deionized water (1 min), and fresh gel matrix (3 min) at high pressure.
Data Analysis:
- The software will generate an electropherogram.
- Identify peaks by comparison with known standards: Aggregate (early migration), Intact mAb, Non-glycosylated Heavy Chain (NGHC), Light Chain (LC).
- Integrate peak areas. Calculate the percentage of each species relative to the total peak area.
Title: CGE-LIF Standard Experimental Workflow
Advanced Protocol: System Suitability and Calibration
Protocol 2: Daily System Suitability Test for Nucleic Acid Analysis
Application: Ensuring system readiness for sensitive oligonucleotide impurity profiling.
Method:
- Prepare a mixture of fluorescently-labeled DNA ladder fragments (e.g., 10 bp, 50 bp, 100 bp, 500 bp) at 0.5 µg/mL each in deionized water.
- Using a 1% polyethylene oxide gel in 1x TBE with 7M urea, perform a separation at 12 kV for 15 minutes (capillary: 40 cm total, 30 cm effective).
- Acceptance Criteria: All peaks must be baseline resolved (Resolution >1.5). The migration time RSD for the 100 bp peak across three replicate runs must be ≤ 1.0%. The signal-to-noise ratio for the 10 bp peak must be > 10:1.
Title: System Suitability Test (SST) Logic Flow
The Scientist's Toolkit: Critical Consumables & Software
Table 3: Essential Toolkit for CGE-LIF Research
| Category | Specific Item | Purpose |
|---|---|---|
| Consumables | Coated Capillaries (e.g., DB-1, eCAP dsDNA) | Provide inert surface for reproducible separation. |
| Replaceable Gel Polymer Kits (various MW ranges) | Enable size-based separation; different kits optimize resolution for proteins, dsDNA, or oligonucleotides. | |
| Fluorescent Labeling Kits (NHS-ester, maleimide) | Enable sensitive LIF detection of proteins at low concentrations. | |
| Standards | Protein/RNA/DNA Ladders (fluorescently labeled) | Essential for system suitability, calibration, and peak identification. |
| Internal Standard (e.g., a unique fluorescent dye) | Corrects for injection variability in quantitative work. | |
| Software | Instrument Control & Acquisition Suite | Manages run parameters, voltage, temperature, and data collection. |
| Advanced Peak Analysis Module | Deconvolutes overlapping peaks, calculates % composition, and performs batch processing for high-throughput labs. |
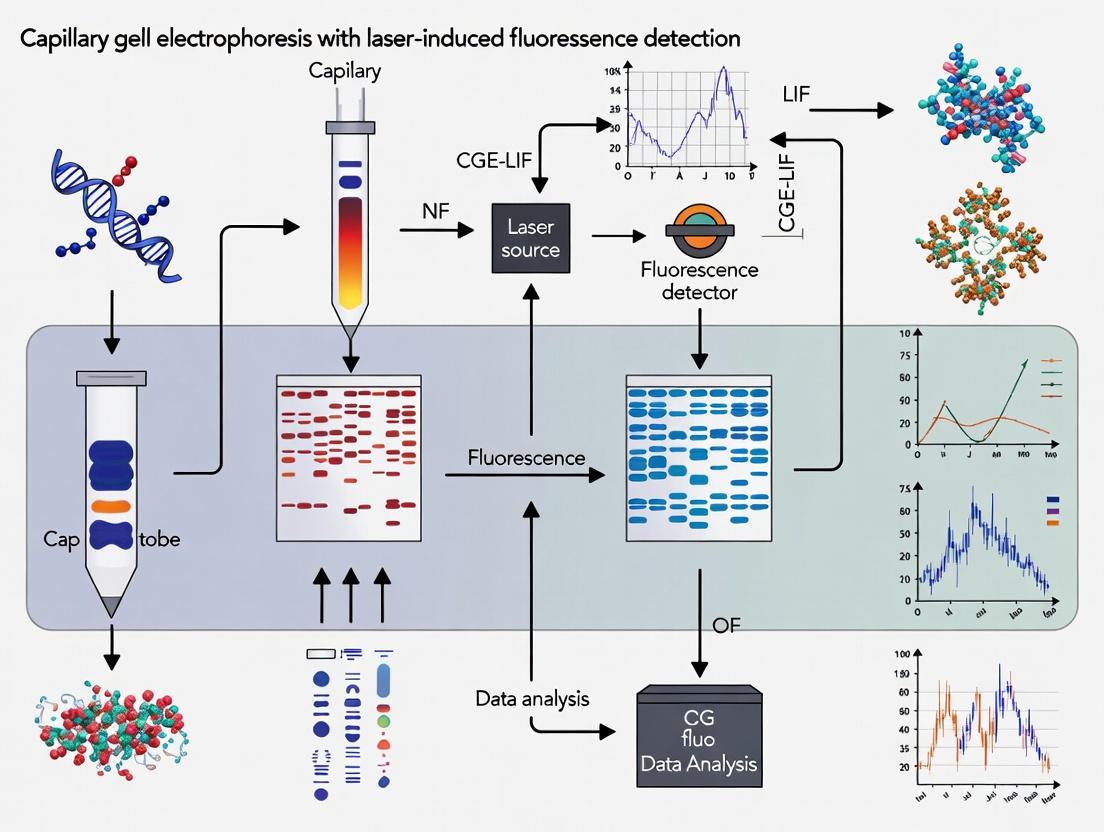
This application note details the core operational principles of capillary gel electrophoresis with laser-induced fluorescence detection (CGE-LIF), a high-sensitivity analytical technique pivotal in biopharmaceutical development for analyzing biomolecules like oligonucleotides, proteins, and carbohydrates. Within the broader thesis of advancing CGE-LIF for next-generation therapeutic characterization, this document explicates the fundamental processes of migration, separation, and detection. It provides validated protocols and essential resources for researchers aiming to implement or optimize CGE-LIF in drug development pipelines.
Capillary Gel Electrophoresis (CGE) is a high-resolution separation technique where charged analytes migrate through a capillary filled with a viscous polymer matrix (gel) under the influence of an electric field. Separation is based on differences in the charge-to-size ratio, with smaller molecules migrating faster than larger ones through the sieving network. Coupled with Laser-Induced Fluorescence (LIF) detection, which offers exceptional sensitivity by exciting fluorescently tagged molecules and measuring emitted light, CGE-LIF is indispensable for analyzing size heterogeneity of DNA fragments, purity of synthetic oligonucleotides (e.g., siRNA, ASOs), and glycosylation patterns of proteins. This note deconstructs the core principles to enable robust method development.
Core Principles & Quantitative Data
Migration: Electrophoretic Mobility
The velocity (v) of an analyte is determined by its electrophoretic mobility (μep) and the applied electric field strength (E). v = μep * E Mobility is influenced by the analyte's charge (q), hydrodynamic radius (r), and buffer viscosity (η): μep = q / (6πηr).
Table 1: Typical Electrophoretic Conditions and Resultant Mobilities for Common Analytes
| Analyte Type | Typical Electric Field (V/cm) | Buffer System | Approx. Mobility (μep, cm²/V·s) |
|---|---|---|---|
| ssDNA (50-mer) | 200-300 | TBE with Denaturing Gel | 3.5 x 10⁻⁴ |
| IgG (Reduced) | 150-200 | SDS-MW Separation Gel | 1.2 x 10⁻⁴ |
| Oligonucleotide (20-mer) | 200-300 | TBE with Denaturing Gel | 4.0 x 10⁻⁴ |
Separation: Sieving Mechanism
The polymer matrix acts as a dynamic sieve. The separation is governed by the Ogston sieving model for smaller molecules and the reptation model for larger polymers.
Table 2: Recommended Gel Polymer Concentrations for Size-Based Separation
| Target Size Range (bp for DNA) | Recommended Polymer (% w/v) | Separation Mechanism Dominance |
|---|---|---|
| 10-500 bp | 6-10% Linear Polyacrylamide | Ogston Sieving |
| 500-5000 bp | 2-4% Linear Polyacrylamide | Reptation |
| Proteins (10-225 kDa) | 3-6% Dextran or PVA (SDS-coated) | Ogston/Reptation Transition |
Detection: LIF Sensitivity
LIF detection involves excitation of a fluorophore by a focused laser (e.g., 488 nm argon-ion). The emitted fluorescence (e.g., 520 nm) is collected by a photomultiplier tube (PMT). Sensitivity is quantified by Limit of Detection (LOD).
Table 3: Typical LIF Detection Performance Metrics
| Fluorophore | Excitation (nm) | Emission (nm) | Typical LOD (Molar) | Common Application |
|---|---|---|---|---|
| FAM | 488 | 520 | 1 x 10⁻¹² | Oligonucleotide Purity |
| Cy5 | 649 | 670 | 5 x 10⁻¹³ | Protein Glycan Profiling |
| SYPRO Ruby | 280, 450 | 610 | 1 x 10⁻¹⁰ | Protein Purity (post-run) |
Experimental Protocols
Protocol 3.1: CGE-LIF for Oligonucleotide Purity and Size Heterogeneity Analysis
Objective: To assess the purity and identify size variants of a synthetic 25-mer antisense oligonucleotide (ASO).
Materials: See "The Scientist's Toolkit" (Section 5). Pre-Run Preparation:
- Capillary Conditioning: Flush a 50 μm ID, 30 cm effective length (40 cm total) bare fused silica capillary with:
- 1.0 M NaOH for 10 min.
- Deionized H₂O for 5 min.
- Separation gel buffer (1x TBE with 7M Urea) for 10 min.
- Gel Loading: Pressure-inject commercially prepared 10% linear polyacrylamide (LPA) gel matrix for 5 min.
- Sample Preparation: Dilute FAM-labeled ASO sample to 100 nM in nuclease-free water. Mix 1:1 with formamide containing 0.01% internal standard (e.g., 15-mer LIZ-labeled oligo). Denature at 95°C for 3 min, then snap-cool on ice.
Run Conditions:
- Instrument: Applied Biosystems 3500 or equivalent CGE-LIF system.
- Detection: LIF, λex=488 nm, λem=520 nm.
- Temperature: 50°C.
- Voltage: +15 kV.
- Injection: Electrokinetic injection at 5 kV for 10 s.
- Run Time: 30 min.
Data Analysis: Integrate peak areas. Calculate percent purity as (Area of Main Peak / Total Area of All Peaks) x 100. Size variants are identified by comparing migration times to an external size ladder (10-100 bp).
Protocol 3.2: CGE-LIF for N-Glycan Profiling of a Monoclonal Antibody
Objective: To separate and quantify released, fluorescently labeled N-glycans from a therapeutic antibody.
Materials: See "The Scientist's Toolkit" (Section 5). Sample Derivatization:
- Release: Release N-glycans from 100 μg of antibody using PNGase F according to manufacturer protocol.
- Labeling: Label purified glycans with APTS (8-aminopyrene-1,3,6-trisulfonic acid) via reductive amination. Incubate glycan sample with 5 mM APTS in 1.2M citric acid and 1M NaBH₃CN at 37°C for 3 hours.
- Clean-up: Remove excess dye using size-exclusion spin columns or ethanol precipitation.
CGE-LIF Analysis:
- Capillary: Use a coated capillary (e.g., polyvinyl alcohol) to minimize electroosmotic flow (EOF).
- Matrix: Pressure-load a commercial carbohydrate separation gel (e.g., 1% dextran in 40mM EACA/0.4% HPMC, pH 9.5).
- Injection: Hydrodynamic injection at 0.5 psi for 10 s.
- Run Conditions: Voltage: +20 kV. Temperature: 25°C. Detection: LIF, λex=488 nm, λem=520 nm.
- Separation: Run for 25 min. Glycans separate by charge and size; sialylated glycans migrate slower.
Quantification: Assign peaks using a glucose ladder unit (GU) value database. Report relative percent abundance of each glycan species (e.g., G0F, G1F, G2F, Man5).
Visualization of Core Workflows
Title: CGE-LIF Standard Analytical Workflow
Title: Interrelated Core Principles of CGE-LIF
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 4: Key Reagent Solutions for CGE-LIF Experiments
| Item | Function & Rationale | Example Product/Chemical |
|---|---|---|
| Linear Polyacrylamide (LPA) Gel | Sieving matrix for high-resolution nucleic acid separation. Minimizes electroosmotic flow (EOF) and analyte adhesion. | Applied Biosystems POP-6, POP-7 |
| Coated Capillary | Suppresses EOF and analyte adsorption to capillary wall, critical for protein/glycan analysis. | DB-1, eCAP Neutral, Polyvinyl Alcohol (PVA) coated |
| Fluorescent Dyes | Covalent or non-covalent tags for LIF detection. Must have high quantum yield and match laser lines. | FAM, Cy5, APTS, SYBR Gold |
| Denaturing Buffer | Contains urea/formamide to keep nucleic acids single-stranded, ensuring separation by size only. | 1x TBE with 7M Urea |
| Size Standard Ladder | Essential for calibrating migration time to analyte size (bp, kDa, GU). | GeneScan 600 LIZ, Dextran Ladder (for glycans) |
| High-Purity Running Buffer | Provides consistent ionic strength and pH for stable current and reproducible mobility. | 1x Tris-Borate-EDTA (TBE), 1x Tris-Glycine-SDS |
| Capillary Regeneration Solutions | Removes residual gel and adsorbed analytes to maintain capillary performance. | 1M NaOH, 0.1M HCl, Deionized Water |
Capillary Gel Electrophoresis with Laser-Induced Fluorescence detection (CGE-LIF) is a cornerstone analytical technique in biopharmaceutical development and characterization. Its unique value proposition for the analysis of proteins, nucleic acids, and complex biologics rests on three interdependent pillars: exceptional sensitivity, high resolution, and robust quantitative power. Within the context of advancing CGE-LIF research, this application note details how optimizing these parameters is critical for applications such as monitoring critical quality attributes (CQAs) of gene therapies, assessing antibody-drug conjugate (ADC) heterogeneity, and quantifying host cell protein (HCP) impurities at ultralow levels.
Data Presentation: CGE-LIF Performance Metrics
Table 1: Comparative Performance of CGE-LIF vs. Other Analytical Techniques
| Performance Metric | CGE-LIF | SDS-PAGE (Coomassie) | SDS-PAGE (Silver Stain) | CE-SDS-UV |
|---|---|---|---|---|
| Detection Sensitivity | Low fM to pM (zeptomole) | ~1-10 ng/band | ~0.1 ng/band | ~0.1-1 µg/mL (femtomole) |
| Dynamic Range | 3-4 orders of magnitude | 1-2 orders of magnitude | <2 orders of magnitude | 2-3 orders of magnitude |
| Resolution (Rs) | ≥1.5 for size variants differing by ≤2% | ~1.0 for variants differing by ~10% | ~1.0 for variants differing by ~10% | ≥1.5 for variants differing by ≤2% |
| Quantitative Precision (%RSD) | <2% (migration time), <5% (peak area) | 10-20% | 15-25% | <2% (migration time), <5-10% (peak area) |
| Sample Consumption | ~10 nL per injection | ~10 µL per lane | ~10 µL per lane | ~10 nL per injection |
| Analysis Time | 20-45 minutes | 2-4 hours (inc. staining) | 3-5 hours (inc. staining) | 20-45 minutes |
| Quantitative Nature | Inherently quantitative (direct detection) | Semi-quantitative (destructive staining) | Semi-quantitative, non-linear (destructive staining) | Inherently quantitative (direct detection) |
Table 2: Key Applications Enabled by CGE-LIF's Core Strengths
| Application Area | Relies on Sensitivity For: | Relies on Resolution For: | Relies on Quantitative Power For: |
|---|---|---|---|
| AAV Capsid Protein Purity | Detecting low-abundance degraded or truncated VP3 proteins (<0.1%). | Separating VP1, VP2, and VP3 isoforms. | Precisely quantifying % full capsid ratio and impurity levels for lot release. |
| ADC Drug-Antibody Ratio (DAR) | Identifying low-abundance DAR species. | Resolving DAR0, DAR2, DAR4, DAR6, etc., populations. | Determining mean DAR and distribution profile for critical PK/PD correlations. |
| mRNA Vaccine Integrity | Detecting trace amounts of fragmented or truncated mRNA. | Separating full-length product from n-1, n+1, and other impurities. | Quantifying % full-length mRNA as a key potency indicator. |
| HCP Analysis | Detecting HCPs at <1 ppm levels in purified drug substance. | Resolving HCPs from product-related variants in complex mixtures. | Accurately reporting ppm levels for impurity clearance validation. |
Experimental Protocols
Protocol 1: Analysis of Adeno-Associated Virus (AAV) Capsid Protein Purity by CGE-LIF
Objective: To quantify the relative abundance of VP1, VP2, and VP3 proteins and detect low-level degradants in purified AAV samples.
Materials: See "The Scientist's Toolkit" below.
Method:
- Sample Denaturation and Labeling:
- Mix 15 µL of AAV sample (∼1x10^12 vp/mL) with 5 µL of 4x Fluorescent Labeling Buffer (containing 1% SDS and 10 mM reducing agent).
- Heat at 70°C for 5 minutes.
- Add 1 µL of 1 mM fluorescent dye (e.g., Chromeo P503) from the Protein Labeling Kit. Vortex and incubate at 70°C for 5 minutes in the dark.
- Quench the reaction with 1 µL of 10x Quenching Buffer.
Instrument Preparation:
- Install a gel-filled capillary (e.g., 50 µm i.d., 20 cm effective length).
- Prime the capillary with Gel Matrix for 3 minutes at high pressure (e.g., 100 psi).
- Set the LIF detector: Excitation = 488 nm, Emission = 520 nm (or filter appropriate for chosen dye).
Separation:
- Inject sample hydrodynamically at 5 psi for 10 seconds.
- Apply a separation voltage of +15 kV at 25°C for 30 minutes.
- Use a zwitterionic CE-SDS running buffer.
Data Analysis:
- Identify peaks based on migration time compared to a protein ladder standard.
- Integrate peak areas for VP1, VP2, and VP3.
- Calculate percentage of each capsid protein: %VPx = (Area of VPx / Total area of VP1+VP2+VP3) * 100.
- Integrate any pre- or post-main peak shoulders to quantify degradants/aggregates.
Protocol 2: Determining Antibody-Drug Conjugate (ADC) Drug-to-Antibody Ratio Distribution
Objective: To resolve and quantify the relative amounts of DAR0, DAR2, DAR4, etc., species in a lysine- or cysteine-conjugated ADC.
Method:
- Sample Reduction and Labeling:
- For cysteine-conjugated ADCs: Dilute ADC to 1 mg/mL in PBS. Mix 10 µL with 10 µL of 2x Non-Reducing Sample Buffer (containing 2% SDS, no DTT/TCEP). Proceed to step 1c.
- For lysine-conjugated ADCs or detailed subunit analysis: Dilute ADC to 1 mg/mL. Mix 10 µL with 10 µL of 2x Reducing Sample Buffer (containing 2% SDS and 50 mM DTT). Heat at 70°C for 5 minutes.
- Add 2 µL of 1 mM fluorescent dye. Vortex and incubate at 70°C for 5 minutes in the dark.
- Quench with 2 µL of 10x Quenching Buffer.
Instrument Preparation: (As per Protocol 1, step 2).
Separation:
- Inject sample at 5 psi for 20 seconds.
- Apply a separation voltage of +15 kV at 25°C for 35 minutes.
Data Analysis:
- Deconvolute the electrophoregram to assign peaks to DAR species based on known incremental molecular weight increases from the drug payload.
- Integrate the area of each DAR peak.
- Calculate percentage distribution: %DARn = (Area of DARn peak / Total integrated area) * 100.
- Calculate weighted average DAR: DARavg = Σ (%DARn * n) / 100.
Visualizations
CGE-LIF Core Workflow: From Injection to Data
UV vs LIF Detection: Impact on Core Analytical Value
The Scientist's Toolkit: Essential Reagents & Materials for CGE-LIF
| Item | Function & Importance |
|---|---|
| Gel-Filled Capillaries | Pre-filled with a sieving polymer matrix (e.g., linear polyacrylamide). Critical for size-based separation. Pre-cast capillaries ensure reproducibility. |
| Fluorescent Protein Labeling Kit | Contains a site-specific dye (e.g., maleimide or NHS-ester), reaction, and quenching buffers. Enables highly sensitive, non-destructive labeling of proteins. |
| CE-SDS Running Buffer (Zwitterionic) | Optimized buffer for SDS-capillary electrophoresis. Reduces electroosmotic flow (EOF) and protein-wall interactions, maximizing resolution. |
| Protein Size Ladder (Fluorescently Labeled) | Mixture of proteins of known molecular weight. Essential for assigning molecular sizes to sample peaks and monitoring system performance. |
| SDS Sample Buffer (Reducing/Non-Reducing) | Contains SDS to denature and impart uniform charge, and may contain DTT/TCEP to reduce disulfide bonds. Defines the analytical context (intact vs. subunit analysis). |
| Capillary Cassette/Cartridge | Holds the capillary, provides alignment for the detection window, and interfaces with temperature control. Essential for robust operation. |
| High-Sensitivity LIF Detector Module | Contains laser light source (e.g., 488 nm, 638 nm) and precise optical filters. The core component enabling femtogram/zeptomole level sensitivity. |
| System Suitability Standard | A well-characterized protein or antibody sample. Run daily to validate the sensitivity, resolution, and migration time precision of the entire CGE-LIF system. |
How to Implement CGE-LIF: Step-by-Step Protocols and Key Applications in Biopharma
Within Capillary Gel Electrophoresis with Laser-Induced Fluorescence detection (CGE-LIF) research, the integrity of analytical data is fundamentally determined by upstream sample preparation. This article details the critical pre-analytical steps of labeling, buffer formulation, and clean-up, providing specific Application Notes and Protocols optimized for high-resolution CGE-LIF analysis of proteins and nucleic acids.
Labeling Strategies for CGE-LIF Sensitivity
Fluorescent labeling is mandatory for LIF detection. The choice of dye and conjugation chemistry must minimize analyte heterogeneity and mobility shifts.
Nucleic Acid Labeling
Intercalating dyes (for dsDNA) and covalent labels (for ssDNA/RNA) are primary strategies.
- Application Note: For dsDNA fragment analysis (e.g., PCR products, NGS libraries), intercalating dyes like SYBR Gold or Ethidium Bromide are added to the separation matrix or sample buffer. They offer universal staining but require post-separation mixing.
- Protocol: Covalent 5'-End Labeling of Oligonucleotides with Alexa Fluor 488
- Materials: Oligonucleotide with 5'-amine modifier, Alexa Fluor 488 NHS ester (succinimidyl ester), 0.1M Sodium Bicarbonate Buffer (pH 8.5), DMSO (anhydrous), Sephadex G-25 spin column.
- Procedure:
- Dissolve the amine-modified oligonucleotide in 50 µL of 0.1M sodium bicarbonate buffer (pH 8.5).
- Dissolve Alexa Fluor 488 NHS ester in anhydrous DMSO to a concentration of 10 mg/mL.
- Add a 10-fold molar excess of the dye solution to the oligonucleotide solution. Mix gently.
- Incubate in the dark at room temperature for 4-6 hours.
- Purify the labeled oligonucleotide using a Sephadex G-25 spin column, pre-equilibrated with TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) or nuclease-free water, to remove excess free dye.
- CGE-LIF Consideration: A single, charge-neutral dye minimizes mobility shift relative to unlabeled size standards.
Protein Labeling
Primary amines (lysine residues, N-terminus) are common targets.
- Application Note: For protein purity and aggregation analysis by CGE-LIF, minimal labeling (≤1 dye/protein) is crucial to avoid creating multiple electrophoretic species.
- Protocol: Minimal Labeling of Proteins with FITC
- Materials: Target protein in amine-free buffer (e.g., 0.1M Sodium Borate, pH 8.5), FITC Isomer I in DMSO, Zeba Spin Desalting Column (7K MWCO).
- Procedure:
- Adjust protein concentration to 1-2 mg/mL in 0.1M sodium borate buffer (pH 8.5).
- Prepare a 10 mg/mL solution of FITC in anhydrous DMSO.
- Add FITC solution to the protein solution at a molar ratio of 5:1 (FITC:Protein). Mix thoroughly.
- Incubate on ice in the dark for 2 hours.
- Terminate the reaction by adding 1M Tris-HCl (pH 6.8) to a final concentration of 50 mM (quenches unreacted FITC).
- Immediately purify the labeled protein using a desalting spin column pre-equilibrated with a CGE-compatible storage buffer (e.g., 25 mM Tris, 192 mM Glycine, pH 8.3) to remove salts and free dye.
Table 1: Common Fluorophores for CGE-LIF Analysis
| Fluorophore | Excitation Max (nm) | Emission Max (nm) | Suitable For | Key Consideration for CGE |
|---|---|---|---|---|
| FITC | 495 | 519 | Proteins | pH-sensitive fluorescence. |
| Alexa Fluor 488 | 495 | 519 | Proteins, Nucleic Acids | More photostable than FITC. |
| Cy5 | 649 | 670 | Nucleic Acids | Common for NGS library QC. |
| SYBR Gold | ~495 (ss) / ~300 (ds) | ~537 | ds/ss Nucleic Acids | Non-covalent, high signal enhancement. |
| 6-FAM | 495 | 520 | Oligonucleotides | Standard for genetic analysis. |
Critical Buffer Formulations
Buffers must maintain analyte stability, minimize capillary wall adsorption, and be compatible with the separation matrix and LIF optics.
Table 2: Essential Buffers for CGE-LIF Sample Preparation
| Buffer Name | Composition | pH | Function in Sample Prep | Notes |
|---|---|---|---|---|
| Denaturing DNA Loading Buffer | 95% Formamide, 10 mM EDTA, 0.025% (w/v) SDS, trace amounts of dextran blue/orange G | 8.0 | For ssDNA/RNA denaturation & density-based loading | Formamide purity is critical. Dextran acts as a neutral pull-up marker. |
| Native Protein Buffer | 25 mM Tris, 192 mM Glycine, 0.1% SDS (optional for CE-SDS) | 8.3 | Protein solubilization & compatibility with CE-SDS | Without SDS for native analysis; with SDS for denatured size analysis. |
| Desalting/Elution Buffer | 10 mM Tris-HCl, 1 mM EDTA (TE) or 0.1% TFA in water/ACN | Varies | Final sample resuspension after clean-up | Low ionic strength is ideal for electrokinetic injection in CGE. |
| Borate-based Conjugation Buffer | 0.1M Sodium Tetraborate (Borax) or Boric Acid-NaOH | 8.5-9.0 | Optimal for amine-reactive labeling (NHS esters) | High pH deprotonates primary amines for efficient conjugation. |
Clean-up Protocols Post-Labeling
Removal of excess dyes, salts, enzymes, and detergents is essential to prevent capillary fouling, injection artifacts, and high background.
Protocol: SPRI Bead-based Clean-up for Nucleic Acids
(Solid Phase Reversible Immobilization)
- Materials: AMPure XP or SPRIselect beads, fresh 80% ethanol, nuclease-free water or TE buffer, magnetic rack.
- Procedure:
- Bring sample (e.g., labeled DNA) to room temperature. Vortex SPRI beads thoroughly.
- Add a calibrated volume of beads to the sample (e.g., 1.8X bead-to-sample ratio for size selection). Pipette mix thoroughly.
- Incubate at room temperature for 5 minutes.
- Place tube on a magnetic rack until the supernatant is clear (~2-5 minutes).
- Carefully remove and discard the supernatant.
- With tube on magnet, add 200 µL of freshly prepared 80% ethanol. Incubate for 30 seconds, then remove ethanol. Repeat for a total of two washes.
- Air-dry beads on magnet for 5-7 minutes until beads appear matte (do not over-dry).
- Remove from magnet. Elute DNA in a low-ionic-strength buffer (e.g., nuclease-free water or TE, pH 8.0) by pipetting. Incubate 2 minutes.
- Place back on magnet and transfer the clean eluate to a new tube.
Protocol: Spin Desalting for Proteins & Small Oligos
- Materials: Zeba or Micro Bio-Spin columns (appropriate MWCO), CGE-compatible storage buffer.
- Procedure:
- Centrifuge the provided storage buffer out of the column (300 x g for 2 minutes for a 7K column).
- Add up to 100 µL of your labeled protein/oligo sample to the center of the resin bed.
- Centrifuge again at the same speed for 2 minutes. The eluate contains the cleaned-up sample.
The Scientist's Toolkit
Table 3: Research Reagent Solutions for CGE-LIF Sample Prep
| Item | Function/Application |
|---|---|
| NHS-Ester Fluorophores (e.g., Alexa Fluor 488 NHS ester) | Covalently labels primary amines (-NH2) on proteins and amine-modified nucleic acids. |
| SYBR Gold Nucleic Acid Gel Stain | Ultrasensitive, non-covalent intercalating dye for ds/ss nucleic acid detection in gels or matrices. |
| AMPure XP / SPRIselect Beads | Magnetic beads for size-selective purification and clean-up of DNA/RNA, removing primers, dyes, and salts. |
| Zeba Spin Desalting Columns | Rapid 7-minute buffer exchange and removal of small molecule contaminants (dyes, salts) for proteins or oligonucleotides. |
| Formamide (Molecular Biology Grade) | High-purity denaturant for preparing nucleic acid samples for denaturing CGE. |
| DTT (Dithiothreitol) / 2-Mercaptoethanol | Reducing agents to break disulfide bonds in proteins for accurate CE-SDS analysis under reducing conditions. |
| High-Purity SDS (Sodium Dodecyl Sulfate) | Anionic detergent for denaturing proteins and providing uniform negative charge in CE-SDS applications. |
| Deionized Water (≥18.2 MΩ·cm) | Essential for preparing all buffers and reagents to prevent ionic contamination that degrades CGE performance. |
Diagrams
Title: Fluorescent Labeling Workflow for CGE-LIF
Title: Pillars of CGE-LIF Sample Preparation
Within Capillary Gel Electrophoresis with Laser-Induced Fluced Detection (CGE-LIF) research, the optimization of running protocols is critical for achieving high-resolution separation of biomolecules, such as oligonucleotides, proteins, and glycans, in drug development. This application note details the core optimization parameters: gel polymer matrix selection, voltage programming, and temperature control, providing researchers with validated protocols to enhance reproducibility and data quality.
Gel Polymer Selection
The sieving matrix is foundational to CGE separation. Selection depends on analyte size, composition, and required resolution.
Table 1: Common Gel Polymers for CGE-LIF
| Polymer Matrix | Typical Concentration | Optimal Size Range | Key Application | Key Property |
|---|---|---|---|---|
| Linear Polyacrylamide (LPA) | 2-6% (w/v) | 10-500 bp (DNA) | DNA fragment analysis, Sanger sequencing | High flexibility, excellent sieving, low viscosity |
| Polyethylene Oxide (PEO) | 1-3% (w/v) | 20-1000 bp, proteins | SDS-coated proteins, some DNA applications | Self-coating, dynamic viscosity, moderate UV absorption |
| Pullulan | 4-10% (w/v) | 100-2000 bp | Large DNA fragments, size heterogeneity | Neutral, hydrophilic, good for charged analytes |
| Polyvinylpyrrolidone (PVP) | 2-8% (w/v) | 50-1000 bp | General-purpose DNA/RNA analysis | Good stability, moderate sieving, often used in kit formulations |
| Commercial LPA-based Kit (e.g., ssDNA 100-R) | As supplied | 10-600 nt | Single-stranded oligonucleotides (therapeutic RNA/DNA) | Optimized for LIF detection, includes necessary additives |
Selection Protocol:
- Define Analyte: Determine size range and nature (ssDNA, dsDNA, protein, glycan).
- Screen Matrices: Test 2-3 candidate polymers at recommended starting concentrations using a standard voltage program (e.g., 15 kV, constant).
- Evaluate Resolution: Calculate resolution (R) between critical peak pairs. R > 1.5 is typically desired for baseline separation.
- Optimize Concentration: For the best candidate, vary polymer concentration (± 1-2%) to maximize resolution for the target size range. Higher concentrations improve resolution for smaller fragments.
Voltage Program Optimization
Voltage programs control migration time, resolution, and heat generation. Step-field or gradient programs can improve resolution across broad size ranges.
Table 2: Voltage Program Parameters for DNA Separation (100-500 bp)
| Program Type | Initial Step | Final Step | Ramp/Transition | Total Run Time | Application Benefit |
|---|---|---|---|---|---|
| Constant Voltage | 15 kV | 15 kV | N/A | ~20 min | Standard analysis, narrow size range. |
| Two-Step Gradient | 10 kV (2 min) | 15 kV (to end) | Instant switch | ~25 min | Improved resolution for larger fragments (>300 bp). |
| Reverse Polarity* | -15 kV (injection) | +15 kV (separation) | Switch after plug mobilization | ~30 min | Analyte stacking, improved peak shape for low-concentration samples. |
| Linear Gradient | 5 kV | 15 kV | Linear over 20 min | ~30 min | Enhanced resolution across very broad size ranges. |
*Requires instrument capable of rapid polarity switching.
Voltage Optimization Protocol:
- Start Constant: Use a constant voltage (e.g., 15 kV) with your selected polymer to establish a baseline.
- Identify Needs: If resolution is poor for larger fragments, implement a low-to-high step gradient.
- Program Setup:
- Injection: Typically 1-5 kV for 10-60 seconds.
- Separation: Begin with a lower voltage (e.g., 10 kV) for 10-20% of the run, then step to a higher voltage (e.g., 15 kV) for the remainder.
- Monitor Joule Heating: Ensure current remains stable; a spike indicates excessive heating.
Temperature Control
Capillary temperature critically impacts viscosity of the polymer matrix, analyte mobility, and intra-capillary convection. Precise control (± 0.1°C) is essential.
Table 3: Temperature Effects and Recommendations
| Analyte Type | Recommended Temperature | Effect of Increased Temperature | Rationale |
|---|---|---|---|
| ssDNA / Oligonucleotides | 50 - 60°C | Reduces secondary structure, improves peak shape and reproducibility. | Denatures stable intra-molecular structures. |
| dsDNA fragments | 30 - 40°C | Decreases buffer viscosity, increases migration speed, may reduce resolution. | Compromise between run time and separation fidelity. |
| SDS-Protein Complexes | 25 - 30°C (constant) | Can cause complex dissociation or excessive viscosity reduction. | Maintains complex integrity and consistent sieving. |
| General | 20°C, 30°C, 40°C | Standard screening temperatures for unknown analytes. |
Temperature Optimization Protocol:
- Initial Run: Use manufacturer's recommended temperature or 30°C.
- Screen Temperatures: Run standard analyte at 20°C, 30°C, and 40°C. Hold all other variables constant.
- Evaluate: Plot migration time vs. temperature (should be linear for well-behaved systems) and resolution of critical pairs vs. temperature.
- Select: Choose the temperature yielding the highest resolution and most stable baseline. For ssDNA, start at 50°C.
Integrated Protocol for Therapeutic Oligonucleotide Analysis (CGE-LIF)
Objective: High-resolution separation of a 20-100 nt single-stranded oligonucleotide sample with impurities.
Research Reagent Solutions & Essential Materials:
| Item | Function/Description |
|---|---|
| Capillary: 50 µm ID, 30 cm effective length (40 cm total), coated (e.g., polyacrylamide) | Separation channel; coating suppresses electroosmotic flow (EOF). |
| Sieving Matrix: Commercial LPA-based gel buffer (e.g., containing urea, TBE) | Provides size-based sieving; urea denatures secondary structure. |
| Running Buffer: Matched to gel buffer (typically same as polymer matrix) | Maintains consistent ionic strength and pH across capillary. |
| Fluorescent Intercalating Dye (e.g., SYBR Gold, To-Pro-3) | Binds nucleic acids for LIF detection (ex: 488 nm/520 nm or 633 nm/670 nm). |
| Size Standard: Fluorescently-labeled oligonucleotide ladder (10-600 nt) | Enables accurate sizing and system performance qualification. |
| Sample Buffer: Formamide with EDTA, or matching gel buffer with dye | Denatures sample and provides conductive medium for electrokinetic injection. |
| Temperature Control System: Peltier-based capillary oven | Provides precise, active temperature control (±0.1°C). |
| High-Voltage Power Supply: Programmable (±30 kV) | Enables implementation of complex voltage gradients. |
Step-by-Step Method:
- Capillary Conditioning: Flush with fresh gel buffer for 3 min at 50 psi.
- Sample/Dye Preparation: Mix 5 µL of oligonucleotide sample with 15 µL of sample buffer containing 0.5 µL of 100X fluorescent dye. Denature at 95°C for 2 min, then place on ice.
- Injection: Electrokinetically inject at 5 kV for 10 seconds.
- Separation: Apply a two-step voltage program:
- Step 1: 10 kV for 3 minutes.
- Step 2: 15 kV for 25 minutes.
- Temperature: Maintain capillary temperature at 50°C.
- Detection: LIF detection with excitation at 488 nm, emission collected at 520 nm.
- Capillary Regeneration: Between runs, flush with gel buffer for 2 min (50 psi). At the end of the day, flush with deionized water for 3 min and air dry for 1 min.
Data Analysis & Validation
- Sizing: Plot log(size) of standards vs. migration time to create a calibration curve. Use a 3rd or 4th-order polynomial fit for best accuracy across broad ranges.
- Resolution Calculation: R = 1.18*(t2 - t1)/(wh1 + wh2), where t is migration time and wh is peak width at half height. Target R > 1.5.
- System Suitability: Daily, run the size standard. Migration time of the principal peak should have a %RSD < 1.0%, and resolution between two specified peaks should be > 1.5.
CGE-LIF Protocol Optimization Workflow
Temperature Effects on CGE Separation
Application Notes
Within the broader thesis on CGE-LIF research, this technique establishes itself as a critical, high-resolution analytical platform for the biopharmaceutical industry. Its unique combination of size-based separation in a gel-filled capillary with the exquisite sensitivity and selectivity of laser-induced fluorescence detection directly addresses the stringent requirements for characterizing therapeutic proteins. For mAbs, CGE-LIF is indispensable for monitoring critical quality attributes (CQAs) such as size variants, including high-molecular-weight (HMW) aggregates and low-molecular-weight (LMW) fragments. For ADCs, the complexity of the analyte—a heterogeneous mixture of antibody conjugates with varying drug-to-antibody ratios (DAR)—demands a technique capable of separating species based on both size and subtle charge differences induced by hydrophobic drug linker attachments. CGE-LIF, particularly when using dyes like Chromeo P503 for pre-column labeling, provides a robust solution for quantifying the distribution of DAR species, free drug linker, and payload-related impurities, which directly impact efficacy and safety.
The quantitative power of CGE-LIF is highlighted in recent studies. The following tables summarize key performance data for mAb and ADC analysis.
Table 1: Representative CGE-LIF Performance Metrics for mAb Purity Analysis
| Analytical Parameter | Typical Value/Range | Comment |
|---|---|---|
| Resolution (Main Peak vs. Fragment) | ≥ 1.5 | Ensures baseline separation for accurate quantitation. |
| Aggregate (HMW) Quantitation Limit | 0.1% - 0.5% | Critical for detecting low-level immunogenic species. |
| Fragment (LMW) Quantitation Limit | 0.5% - 1.0% | Monitors protein degradation. |
| % Relative Standard Deviation (RSD) for Main Peak Area | < 2.0% | Demonstrates high precision of the method. |
| Migration Time RSD | < 1.0% | Indicates excellent run-to-run reproducibility. |
Table 2: CGE-LIF Analysis of a Model ADC: DAR Distribution and Impurity Profile
| Species Resolved | Relative Migration Time (Normalized) | Typical Percentage (%) | Significance |
|---|---|---|---|
| High-Molecular-Weight Aggregate (HMW) | 0.92 - 0.95 | 0.2 - 5.0 | Potential immunogenicity risk. |
| DAR 4 | 0.98 | 15 - 25 | Often a target peak for optimal efficacy/toxicity balance. |
| DAR 2 (Main Peak) | 1.00 (reference) | 40 - 60 | Major species in a typical distribution. |
| DAR 0 (Naked mAb) | 1.03 | 5 - 15 | Unconjugated antibody, lower potency. |
| Free Drug Linker / Payload | 1.10 - 1.20 | < 2.0 | Small molecule impurity, safety concern. |
Experimental Protocols
Protocol 1: Sample Preparation and Fluorescent Labeling for mAb/ADC Analysis This protocol details the derivatization of protein samples with Chromeo P503 dye to enable LIF detection.
- Reagent Preparation: Prepare a 10 mM stock solution of Chromeo P503 in anhydrous dimethylformamide (DMF). Prepare a 100 mM borate buffer, pH 8.5.
- Sample Dilution: Dilute the mAb or ADC sample to a concentration of 1 mg/mL using the borate buffer.
- Labeling Reaction: Mix 10 µL of the 1 mg/mL protein sample with 2 µL of the 10 mM Chromeo P503 dye solution. Vortex gently.
- Incubation: Protect the reaction mixture from light and incubate at room temperature for 30 minutes.
- Reaction Quenching: Add 2 µL of a 1.5 M hydroxylamine solution (pH 8.5) to quench the reaction. Mix well and incubate for 15 minutes at room temperature, protected from light.
- Sample Dilution for Analysis: Dilute the labeled sample 1:10 with deionized water prior to CGE-LIF injection.
Protocol 2: CGE-LIF Instrumental Method for mAb/ADC Size Variant Analysis This method is optimized for use with a Beckman PA 800 Plus or equivalent system equipped with a LIF detector using a 488 nm excitation laser and a 520 nm emission filter.
- Capillary Conditioning: Flush a new or used bare-fused silica capillary (50 cm total length, 40 cm to detector, 50 µm inner diameter) with 0.1 M HCl for 5 min, deionized water for 3 min, and then SDS gel running buffer for 10 min.
- Gel Matrix Preparation: Prepare the SDS-MW gel solution according to the manufacturer's instructions (e.g., Beckman Coulter SDS-MW kit). Centrifuge the gel solution at 10,000 x g for 10 minutes to degas and remove particulates.
- Capillary Filling: Pressure-fill the capillary with the fresh SDS-MW gel matrix for 10 minutes.
- Instrument Parameters:
- Separation Voltage: +15 kV.
- Temperature: 25°C.
- Injection: Electrokinetic injection at 5 kV for 20 seconds.
- Detection: LIF, excitation 488 nm, emission 520 nm.
- Run Sequence: Include a system suitability standard (e.g., a characterized mAb or protein ladder) and a buffer blank at the beginning of each sequence. Inject samples in duplicate.
- Data Analysis: Use the instrument software (e.g., 32 Karat) to integrate peaks. Identify species based on relative migration time (RMT) compared to the main peak or a ladder. Calculate percentage areas for each peak relative to the total integrated area.
Mandatory Visualization
Diagram 1: CGE-LIF Workflow for ADC Analysis
Diagram 2: ADC Species Resolved by CGE-LIF
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for CGE-LIF of mAbs/ADCs
| Item | Function / Purpose | Typical Example / Specification |
|---|---|---|
| Fluorescent Dye | Covalently labels primary amines (lysine) on proteins for sensitive LIF detection. | Chromeo P503, Alexa Fluor 488 NHS ester. |
| SDS-MW Gel Matrix | A replaceable, polymer-based sieving matrix for size-based separation (SDS-CGE). | Beckman Coulter SDS-MW Kit (Part No. 390953). |
| SDS Running Buffer | Provides consistent ionic strength and SDS for maintaining protein charge and separation. | Tris-Tricine-SDS buffer, pH ~7.6. |
| Capillary | The separation channel. Bare fused silica is standard. | Bare fused silica, 50 µm ID, 30-50 cm effective length. |
| Capillary Regeneration Solutions | For cleaning and conditioning the capillary between runs to ensure reproducibility. | 0.1 M HCl, 0.1 M NaOH, Deionized Water. |
| Internal/System Suitability Standard | Validates instrument performance and migration time consistency. | Fluorescently-labeled protein ladder or a well-characterized mAb standard. |
| Hydroxylamine Quenching Solution | Stops the fluorescent labeling reaction by reacting with unbound dye. | 1.5 M Hydroxylamine, pH adjusted to 8.5. |
Capillary gel electrophoresis with laser-induced fluorescence detection (CGE-LIF) is a cornerstone analytical technique in the development of nucleic acid therapeutics. Within the broader thesis of advancing CGE-LIF methodologies, this application note addresses the critical challenge of characterizing size heterogeneity—a key quality attribute for oligonucleotides (OGNs) and messenger RNA (mRNA). Precise determination of full-length product, shortmers, longmers, and degradation fragments is non-negotiable for ensuring therapeutic efficacy and safety. This protocol details optimized CGE-LIF methods for high-resolution separation and sensitive quantification of these critical impurities.
Key Analytical Performance Data
The following table summarizes typical performance metrics for CGE-LIF analysis of OGNs and mRNA using commercially available gel matrices and optimized protocols.
Table 1: CGE-LIF Performance Metrics for Nucleic Acid Therapeutics
| Parameter | Antisense Oligonucleotides (ASO, ~20 nt) | siRNA (Duplex, 21-23 bp) | mRNA (1000-5000 nt) |
|---|---|---|---|
| Separation Matrix | ssDNA Gel Buffer (e.g., POP6/7) | dsDNA Gel Buffer (e.g., POP6/7) | RNA Gel Buffer (e.g., POP6) |
| Typical Capillary | 50 µm ID, 30-50 cm effective length | 50 µm ID, 30-50 cm effective length | 50 µm ID, 30-50 cm effective length |
| Resolution (Rs) | ≥ 1.5 between n and n-1 | ≥ 2.0 between single strands | Capable of resolving Δ ~50 nt fragments |
| Limit of Detection (LOD) | ~0.1 ng/µL (Sybr Gold) | ~0.2 ng/µL (Sybr Gold) | ~0.5 ng/µL (RiboGreen) |
| Migration Time RSD | < 0.5% | < 0.8% | < 1.5% |
| Area % RSD | < 2.0% | < 3.0% | < 5.0% |
| Primary QC Readout | Full-length purity (% FLP) | Duplex purity, guide/passenger strand ratio | Integrity (IVT product vs. truncated), purity from dsRNA |
Detailed Experimental Protocols
Protocol 1: Purity Analysis of Single-Stranded Oligonucleotides
Objective: Quantify full-length product and related shortmer impurities. Materials: See "The Scientist's Toolkit" below. Procedure:
- Sample Preparation: Dilute the OGN sample in nuclease-free water to a final concentration of 50-100 ng/µL. Add an internal size standard (e.g., 10-30 nt ladder) at 10% (v/v) if absolute mobility confirmation is required.
- Fluorescent Labeling: For LIF detection, intercalating dyes are used in the run buffer. For pre-labeling, follow dye manufacturer's protocol for covalent modification (e.g., 5'-FAM).
- Instrument Setup: Install a bare fused silica capillary (50 µm ID, 30 cm effective length). Use a commercial ssDNA analysis gel buffer (e.g., POP-6 or -7). Set the instrument temperature to 35°C.
- Run Conditions: Apply a voltage of 12-15 kV for 20-30 minutes. Use a pressure injection (0.5 psi for 10-20 seconds) or electrokinetic injection (5 kV for 10 seconds).
- Data Analysis: Identify peaks by comparison to a size ladder. Calculate the area percentage of the full-length peak relative to the total integrated area of all oligonucleotide-related peaks to determine % full-length product.
Protocol 2: Integrity Analysis of mRNA Therapeutics
Objective: Assess mRNA intactness and detect degradation fragments. Materials: See "The Scientist's Toolkit" below. Procedure:
- Sample Denaturation: Dilute mRNA to ~100 ng/µL in nuclease-free water. Heat denature at 70°C for 3 minutes, then immediately place on ice for 2 minutes.
- Gel Matrix Preparation: Use a commercial RNA-specific gel matrix or a high-sensitivity DNA gel matrix validated for RNA. Ensure the matrix contains a fluorescent intercalating dye (e.g., proprietary dye in the buffer) or add it according to the manufacturer's instructions.
- Instrument Setup: Install a capillary (50 µm ID, 30 cm effective length) and fill with the gel matrix. Set the instrument temperature to 40-50°C to minimize secondary structure.
- Run Conditions: Apply a voltage of 10-12 kV for 30-40 minutes. Use a pressure injection (0.5 psi for 15-30 seconds).
- Data Analysis: The main peak corresponds to the intact mRNA. Earlier migrating peaks are truncated or degraded species. Calculate the percentage of the main peak area relative to the total area. The ratio of the main peak area to the total area indicates the percentage of intact mRNA.
Visualization of Workflows
CGE-LIF Analysis Workflow for Nucleic Acids
mRNA Synthesis and CGE-LIF QC Pathway
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for CGE-LIF Analysis of OGNs/mRNA
| Item | Function & Importance | Example Product Types |
|---|---|---|
| Commercial Gel Matrix | Provides the sieving network for size-based separation. Critical for resolution and reproducibility. | ssDNA/RNA Gel Buffer (e.g., POP-6, -7), dsDNA 1000 Gel Buffer. |
| Intercalating Fluorescent Dye | Binds nucleic acids for sensitive LIF detection. Must be compatible with gel matrix and excitation laser. | SYBR Gold, YO-PRO-1, proprietary dyes in kit buffers. |
| Nuclease-Free Water | Prevents sample degradation during dilution and preparation. Essential for accurate analysis of RNA. | USP-grade, DEPC-treated, or 0.1 µm filtered water. |
| Internal/Oligo Size Standard | Calibrates migration time to nucleotide length, enabling identification of impurity sizes. | Fluorescently labeled oligonucleotide ladders (e.g., 10-150 nt). |
| Capillary Conditioning Solutions | Maintains capillary surface consistency, ensuring stable electroosmotic flow and migration times. | 0.1M NaOH, 0.1M HCl, deionized water, gel matrix. |
| Denaturing Agent (for RNA) | Disrupts secondary structure in mRNA to ensure separation is based on length, not conformation. | Formamide (deionized) or high temperature (50-70°C). |
Application Note: Charge Variant Analysis of Monoclonal Antibodies by CGE-LIF
Within the broader thesis on CGE-LIF research, precise characterization of therapeutic protein charge heterogeneity is critical for ensuring efficacy and safety. Charge variants, arising from post-translational modifications, can impact stability, pharmacokinetics, and immunogenicity.
Table 1: Charge Variant Distribution of a Model mAb (Trastuzumab Biosimilar) by CGE-LIF
| Variant | Relative Percentage (%) | Migration Time (min) | Peak Area (RFU) |
|---|---|---|---|
| Acidic 1 | 15.2 ± 0.3 | 8.45 | 15245 |
| Acidic 2 | 10.8 ± 0.2 | 9.10 | 10802 |
| Main Peak | 68.5 ± 0.5 | 10.05 | 68495 |
| Basic 1 | 4.1 ± 0.1 | 10.92 | 4101 |
| Basic 2 | 1.4 ± 0.1 | 11.60 | 1405 |
Detailed Protocol: CGE-LIF for mAb Charge Variants
Principle: Separates charge variants based on differential migration in a coated capillary under an electric field, with LIF detection.
Materials & Reagents:
- CGE-LIF System: PA 800 Plus Pharmaceutical Analysis System (or equivalent) with LIF detector (488 nm excitation/520 nm emission).
- Capillary: Fused silica, 50 µm i.d., 30.2 cm total length (20 cm effective), coated for electroosmotic flow suppression.
- Run Buffer: 100 mM ε-aminocaproic acid, 0.1% hydroxypropylmethylcellulose (HPMC), pH 5.0.
- Sample Buffer: 25 mM sodium phosphate, pH 7.0.
- Internal Standard: Fluorescently labeled pI marker.
- mAb Sample: 1 mg/mL in sample buffer.
Procedure:
- Capillary Conditioning: Flush with 0.1 M HCl (5 min), deionized water (5 min), run buffer (10 min).
- Sample Preparation: Dilute mAb to 1 mg/mL in sample buffer. Mix with internal standard (1:10 v/v).
- Injection: Hydrodynamic injection at 0.5 psi for 10 seconds.
- Separation: Apply voltage of 15 kV for 20 minutes at 25°C.
- Detection: LIF detection with 488 nm laser.
- Analysis: Integrate peaks and calculate relative percentages using proprietary software.
Application Note: Glycan Profiling of Biotherapeutics by CGE-LIF
Glycosylation is a critical quality attribute. This application note details a high-sensitivity CGE-LIF method for profiling released N-glycans, framed within the thesis goal of developing ultra-sensitive biopharmaceutical characterization tools.
Table 2: N-Glycan Profile of a Model IgG1 Antibody
| Glycan Species | Abbreviation | Relative Abundance (%) | Gu (Glucose Units) |
|---|---|---|---|
| G0F | FA2 | 32.5 ± 1.2 | 7.45 |
| G1F | A2G1F | 25.1 ± 0.8 | 7.85 |
| G2F | A2G2F | 18.7 ± 0.7 | 8.24 |
| G0 | A2 | 12.4 ± 0.5 | 6.92 |
| G1 | A2G1 | 5.8 ± 0.3 | 7.33 |
| Man5 | M5 | 3.2 ± 0.2 | 6.51 |
| Sialylated Glycans (Total) | - | 2.3 ± 0.2 | 8.6-9.2 |
Detailed Protocol: CGE-LIF for Released N-Glycan Profiling
Principle: Glycans are released, labeled with a charged fluorescent dye (APTS), and separated by size via sieving electrophoresis.
Materials & Reagents:
- CGE-LIF System: As above.
- Capillary: 50 µm i.d., 40 cm total length (30 cm effective), filled with gel separation matrix.
- Separation Matrix: Commercial glycan separation gel buffer (e.g., Tris-Borate-EDTA with linear polymer).
- APTS Labeling Kit: Contains 8-aminopyrene-1,3,6-trisulfonic acid.
- Glycan Release Kit: PNGase F enzyme.
- Glucose Ladder Standard: APTS-labeled maltooligosaccharides for Gu calibration.
Procedure:
- N-Glycan Release: Denature 50 µg mAb, incubate with PNGase F at 37°C for 3 hours.
- Glycan Labeling: Desalt released glycans. Incubate with APTS in 15% acetic acid and 1M NaBH3CN at 55°C for 2 hours. Quench with water.
- Sample Clean-up: Remove excess dye using purification cartridges.
- Sample Dilution: Dilute labeled glycans 1:10 in deionized water.
- Capillary Conditioning: Flush with separation matrix for 5 min.
- Injection: Electrokinetic injection at 5 kV for 10 seconds.
- Separation: Apply voltage of 20 kV for 25 minutes at 25°C.
- Detection & Analysis: LIF detection. Assign peaks using Gu values from the co-injected ladder. Integrate and calculate relative percentages.
Application Note: Plasmid DNA Homogeneity and Topology Analysis by CGE-LIF
In gene therapy and vaccine development, plasmid DNA (pDNA) topology must be rigorously controlled. This note describes a CGE-LIF method to quantify pDNA isoforms, supporting the thesis's focus on nucleic acid analysis.
Table 3: Topology Analysis of a 5 kbp Plasmid DNA Preparation
| Topoisoform | Relative Percentage (%) | Migration Time (min) | Notes |
|---|---|---|---|
| Supercoiled (SC) | 78.5 ± 2.1 | 12.1 | Desired active form |
| Open Circular (OC) | 15.3 ± 1.5 | 13.8 | Nicked form |
| Linear | 4.2 ± 0.8 | 14.5 | Double-strand break |
| Dimer/Multimer | 2.0 ± 0.5 | 11.3 | Higher-order complex |
Detailed Protocol: CGE-LIF for pDNA Topology Analysis
Principle: Different pDNA topoisomers are separated based on their hydrodynamic size and charge density in a gel-filled capillary.
Materials & Reagents:
- CGE-LIF System: As above, with intercalating dye (e.g., YO-PRO-1) in sieving matrix for on-column staining.
- Capillary: 100 µm i.d., 30 cm total length (20 cm effective).
- Sieving Matrix: 1% (w/v) hydroxyethyl cellulose in 1x TBE buffer with 1 µM YO-PRO-1 dye.
- Running Buffer: 1x Tris-Borate-EDTA (TBE), pH 8.3.
- pDNA Sample: 50 ng/µL in TE buffer or nuclease-free water.
- Size Standard: DNA ladder (0.1-10 kbp).
Procedure:
- Matrix Preparation: Dissolve HEC in TBE, filter, and add intercalating dye.
- Capillary Filling: Pressure flush capillary with sieving matrix for 10 min.
- Sample Preparation: Dilute pDNA to 50 ng/µL.
- Injection: Electrokinetic injection at 5 kV for 10 seconds.
- Separation: Apply voltage of 10 kV at 25°C for 30 minutes. Reverse polarity (cathode at detector side).
- Detection: LIF detection (excitation 488 nm, emission 520 nm).
- Analysis: Identify peaks by comparison with standards. Quantify area percentages for each topoisoform.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 4: Key Reagents and Materials for CGE-LIF Applications
| Reagent/Material | Function / Purpose | Example Application |
|---|---|---|
| Coated Capillaries (e.g., neutral coating) | Suppresses electroosmotic flow (EOF), essential for high-resolution charge-based separations. | Charge Variant Analysis |
| High-Performance Gel Matrix (e.g., linear polyacrylamide) | Acts as a molecular sieve, separating analytes by size. | Glycan Profiling, pDNA Analysis |
| Fluorescent Label (APTS) | Imparts charge and enables highly sensitive LIF detection of neutral sugars. | Glycan Profiling |
| Intercalating Dye (YO-PRO-1) | Binds stoichiometrically to dsDNA, enabling sensitive, quantitative detection. | Plasmid DNA Characterization |
| pI Markers (Fluorescent) | Serve as internal standards for migration time normalization in charge analysis. | Charge Variant Analysis |
| Glucose Unit (GU) Ladder (APTS-labeled) | Calibrates migration time to standard Glucose Units for glycan identification. | Glycan Profiling |
| DNA Size Ladder (0.1-10 kbp) | Used to calibrate migration time for size estimation in nucleic acid separations. | Plasmid DNA Characterization |
| Highly Purified Sieving Polymers (HEC, HPMC) | Provides reproducible viscosity and sieving properties for separation matrix. | All Applications |
Visualization: Experimental Workflows and Relationships
Title: CGE-LIF Workflow for Charge Variant Analysis
Title: CGE-LIF N-Glycan Profiling Workflow
Title: CGE-LIF Plasmid DNA Topology Analysis Workflow
Title: CGE-LIF Applications in Biopharma Thesis Context
Solving Common CGE-LIF Problems: Peak Artifacts, Sensitivity Loss, and Method Robustness
Within Capillary Gel Electrophoresis with Laser-Induced Fluorescence detection (CGE-LIF) research, high-resolution separation of biomolecules like oligonucleotides, proteins, or antibodies is paramount. Peak anomalies directly impact data integrity, quantitation, and the validity of conclusions in drug development. This application note details the diagnosis and resolution of common electrophoregram artifacts, framed within the rigorous demands of biopharmaceutical analysis.
Common Anomalies: Diagnosis and Resolution
The following table summarizes the root causes and corrective actions for the four primary peak anomalies encountered in CGE-LIF.
Table 1: Diagnosis and Resolution of Common CGE-LIF Peak Anomalies
| Anomaly | Primary Diagnostic Clues | Probable Root Cause(s) | Recommended Corrective Actions & Protocols |
|---|---|---|---|
| Broad Peaks | Increased peak width at half height (PWHH); loss of resolution; consistent across samples. | 1. Capillary Overload: Sample concentration too high.2. Temperature Instability: Fluctuations in cartridge/capillary temperature.3. Buffer Depletion: Old or contaminated gel matrix/running buffer. | Protocol 1: Sample Load Optimization. Perform a dilution series (e.g., 1:2, 1:5, 1:10) of the stock sample. Inject using fixed parameters (e.g., 5 kV for 10 sec). The dilution yielding symmetric peaks with minimal PWHH increase is optimal. |
| Shoulders | Asymmetry on the leading or trailing edge of a main peak; may indicate co-migration. | 1. Incomplete Denaturation: Secondary structure in nucleic acids or proteins.2. Matrix Interaction: Sample interacting with capillary wall or gel matrix.3. Buffer Incompatibility: Sample solvent ionic strength/pH mismatched with run buffer. | Protocol 2: Enhanced Denaturation. For oligonucleotides, heat sample to 90°C for 2 mins in formamide-based denaturant, then immediately place on ice for 5 mins before injection. Ensure run buffer contains 7M urea. |
| Extra Peaks | Unanticipated peaks; may be system peaks, degradation products, or contaminants. | 1. Sample Degradation: Enzymatic or chemical breakdown (e.g., hydrolysis, deamidation).2. Carryover: Incomplete capillary wash from previous run.3. Buffer/Reagent Artifacts: Impurities in reagents or fluorescent dye. | Protocol 3: Forced Degradation Study. Incubate a sample aliquot at stressed conditions (e.g., 40°C, 75% RH for 24-72 hrs). Compare to control stored at -80°C. If extra peaks increase with stress, confirm degradation. Implement stringent wash cycles (e.g., 3 min flush with 0.1M NaOH, followed by 3 min gel buffer). |
| Noisy Baseline | High-frequency signal fluctuations; increased baseline RMS noise. | 1. Detection Issues: Unstable laser (power fluctuations), dirty optics, or PMT voltage too high.2. Electrical Noise: Grounding issues or voltage fluctuations.3. Contamination: Particulates in buffer or capillary. | Protocol 4: Laser & Optical Path Check. Monitor laser power output for stability. Perform instrument's optical alignment test. Clean detection window with methanol and lint-free wipe. Filter all buffers and gel matrix through a 0.45 µm filter before use. |
Experimental Workflow for Anomaly Investigation
The logical process for systematically troubleshooting peak anomalies in a CGE-LIF assay is outlined below.
Diagram Title: CGE-LIF Peak Anomaly Troubleshooting Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Robust CGE-LIF Analysis
| Item | Function in CGE-LIF | Critical Notes |
|---|---|---|
| Performance Optimized Polymer (POP) Gel Matrix | Sieving matrix for size-based separation. Provides reproducible migration times and high resolution. | Must be compatible with LIF detection. Store at 4°C, filter after thawing. Lot-to-lot consistency is critical. |
| Fluorescent Intercalating Dye (e.g., SYBR Gold) | Binds nucleic acids non-covalently, enabling high-sensitivity LIF detection. | Must have stable fluorescence in gel matrix. Optimize concentration to avoid quenching or altered mobility. |
| DNA Size Ladder | Essential for system suitability and assigning sizes to unknown sample peaks. | Should cover expected size range. Use same dye and matrix as samples for accurate calibration. |
| Capillary Cartridge (e.g., 50 µm ID, 30 cm effective length) | The separation channel. Coated capillaries prevent electroosmotic flow (EOF) and analyte adsorption. | Regular conditioning (NaOH flush) maintains wall integrity. Check for bubbles or window cracks. |
| Denaturing Buffer (7-8 M Urea with EDTA) | Maintains nucleic acids in a single-stranded, linear state during separation. | Fresh preparation is key; old urea degrades to cyanate, causing carbamylation and artifactual peaks. |
| High-Purity Water (HPLC/LC-MS Grade) | Solvent for all buffers, gels, and sample preparation. | Ionic/organic contaminants directly cause noisy baselines and extra peaks. |
Methodical diagnosis of CGE-LIF peak anomalies is foundational to generating reliable data for critical decision-making in biopharmaceutical development. By integrating the structured workflows, protocols, and reagent management practices outlined here, researchers can enhance assay robustness, ensure data quality, and accelerate therapeutic development timelines.
Within Capillary Gel Electrophoresis with Laser-Induced Fluorescence detection (CGE-LIF) research, achieving maximal sensitivity is paramount for applications such as oligonucleotide impurity profiling, glycoprotein analysis, and single-cell biomolecule detection. Sensitivity is a multiplicative function of several key factors: the efficiency of the fluorescent labeling reaction, the excitation power of the laser, and the precise alignment of the optical detection system. Deficiencies in any one area can lead to suboptimal signal-to-noise ratios, reduced limits of detection, and unreliable quantitative data. This application note provides a structured troubleshooting guide and detailed protocols for systematically optimizing these three critical parameters.
Table 1: Impact of Labeling Efficiency on Signal Intensity
| Labeling Reagent | Optimal Dye-to-Protein (D/P) Ratio | Typical Efficiency Range | Impact on CGE-LIF Peak Height (vs. 50% efficiency) |
|---|---|---|---|
| NHS-ester Cy5 | 3.0 - 5.0 | 60 - 85% | +120% to +180% |
| Alexa Fluor 488 | 4.0 - 6.0 | 70 - 90% | +140% to +200% |
| ATTO 550 | 2.5 - 4.0 | 65 - 88% | +130% to +190% |
Table 2: Laser Power Optimization for Common Fluorophores
| Fluorophore | Excitation Wavelength (nm) | Recommended Laser Power Range (mW) | Signal-to-Noise Ratio at Optimal Power | Risk of Photobleaching Beyond |
|---|---|---|---|---|
| FITC | 488 | 3 - 8 | 150:1 | 10 mW |
| Cy3 | 532 | 5 - 12 | 220:1 | 15 mW |
| Alexa 647 | 635 | 7 - 15 | 300:1 | 20 mW |
Table 3: Detector Alignment Sensitivity Metrics
| Alignment Parameter | Acceptable Tolerance | Measured Impact on CV of Migration Time | Impact on Peak Area Reproducibility (CV) |
|---|---|---|---|
| Capillary Vertical Position | ± 10 µm | Increases from 0.5% to 2.1% | Increases from 1.2% to 8.5% |
| Laser Focus Spot Size | < 50 µm diameter | Minimal impact on CV | Critical for sensitivity; optimal = 30 µm |
| PMT Voltage | ± 50 V | No impact | Directly proportional; CV <2% at set V |
Experimental Protocols
Protocol 3.1: Optimizing Fluorophore Labeling Efficiency for Oligonucleotides
Objective: To achieve consistent, high-efficiency labeling of oligonucleotides with a 5'-amine modifier using a succinimidyl ester dye. Materials: 5'-amine-modified oligonucleotide, NHS-ester dye (e.g., Cy5), anhydrous DMSO, 0.1 M sodium bicarbonate buffer (pH 8.5), desalting spin column, spectrophotometer.
- Preparation: Dissolve the oligonucleotide in nuclease-free water to 1 mM. Dissolve the NHS-ester dye in anhydrous DMSO to 10 mM.
- Reaction: Mix oligonucleotide with 0.1 M sodium bicarbonate buffer (pH 8.5). Add the dye solution at a 10:1 molar ratio (dye:oligonucleotide). Vortex gently.
- Incubation: React in the dark at room temperature for 2 hours.
- Purification: Purify the labeled oligonucleotide using a desalting spin column according to the manufacturer's instructions to remove unconjugated dye.
- Quantification: Measure absorbance at the oligonucleotide's λmax (260 nm) and the dye's λmax (e.g., 650 nm for Cy5). Calculate labeling efficiency (D/P ratio) using the formula: D/P = (Adye / εdye) / (A260 - (Adye * CF)) / ε_oligo), where CF is the dye's correction factor at 260 nm.
- CGE-LIF Analysis: Inject the purified product and analyze. Target a D/P ratio of 0.9 - 1.1 for single labeling.
Protocol 3.2: Systematic Laser Power and Detector Alignment Calibration
Objective: To determine the optimal laser power and validate detector alignment for maximum signal-to-noise ratio (SNR). Materials: CGE-LIF system with adjustable laser, alignment standard (e.g., 100 nM fluorescein solution), test sample (e.g., labeled oligonucleotide at known concentration), capillary cartridge.
- Initial Alignment with Standard:
- Install a capillary filled with run buffer.
- Introduce the fluorescein alignment standard by pressure injection.
- Set laser power to 5% of maximum and PMT voltage to a medium level (e.g., 500 V).
- Using the instrument's alignment software or manual controls, adjust the capillary's position in the detection window and the focusing optics until the real-time signal is maximized. This ensures the laser is focused on the capillary bore and the emitted light is efficiently collected.
- Laser Power Sweep:
- Inject a consistent amount of a low-concentration test sample.
- Run separations at increasing laser power levels (e.g., 10%, 25%, 50%, 75%, 100% of max).
- Record the peak height and baseline noise for a target peak at each power.
- Calculate SNR (Peak Height / Baseline Noise) for each run.
- Data Analysis: Plot SNR vs. Laser Power. Identify the power level where SNR plateaus or begins to decrease due to increased photobleaching or background noise. This is the optimal operational power.
- PMT Voltage Verification: At the optimal laser power, run the sample at different PMT voltages. Ensure the signal response is linear with voltage without saturating the detector.
Visualization
Diagram Title: Sensitivity Troubleshooting Workflow
Diagram Title: LIF Optical Detection Pathway
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Materials for CGE-LIF Sensitivity Optimization
| Item | Function & Role in Optimization |
|---|---|
| NHS-Ester Fluorescent Dyes (e.g., Cy series, Alexa Fluor) | Reactive dyes for covalent labeling of amines on proteins, oligonucleotides, or carbohydrates. High purity is critical for maximizing labeling efficiency (D/P ratio). |
| Anhydrous Dimethyl Sulfoxide (DMSO) | Anhydrous solvent for dissolving and stabilizing NHS-ester dyes. Prevents dye hydrolysis prior to reaction, ensuring consistent labeling. |
| Desalting Spin Columns (Size Exclusion) | Rapid purification of labeled biomolecules from excess free dye. Essential for accurate D/P calculation and preventing background interference in CGE. |
| Fluorescent Alignment Standard (e.g., 100 nM Fluorescein) | A stable, bright fluorophore solution used to optically align the laser focal point with the capillary and maximize light collection by the detector. |
| Capillaries with Custom Detection Window | Fused-silica capillaries with a carefully created, optically clear window. Window quality directly impacts background scatter and signal collection efficiency. |
| Validated CGE Separation Gel Buffer | A proprietary sieving matrix (e.g., POP-7 for DNA) optimized for resolution and compatibility with LIF detection, minimizing fluorescent background. |
Application Notes and Protocols
1. Introduction Within Capillary Gel Electrophoresis with Laser-Induced Fluced detection (CGE-LIF) research, achieving high reproducibility is paramount for the analysis of biotherapeutics, gene therapies, and other critical biomolecules. The analytical thesis posits that systematic control of three principal variables—capillary coating stability, gel matrix lot consistency, and electrokinetic injection precision—is the cornerstone of robust and transferrable CGE-LIF methods. This document provides detailed protocols and application notes to address these specific challenges.
2. Core Challenges and Quantitative Data Summary Key sources of variability are quantified in the following tables.
Table 1: Impact of Capillary Coating Degradation on Migration Time Reproducibility
| Coating Type | Initial Efficiency (Plates/m) | Efficiency after 50 Runs (Plates/m) | %RSD Migration Time (Runs 1-20) | %RSD Migration Time (Runs 30-50) |
|---|---|---|---|---|
| Polyacrylamide (Covalent) | 800,000 | 750,000 | 0.8% | 1.5% |
| Polyvinyl Alcohol (Dynamic) | 750,000 | 500,000 | 1.2% | 4.8% |
| Hydrophilic Polymer (Stable) | 820,000 | 810,000 | 0.7% | 0.9% |
Table 2: Gel Matrix Lot Variability Analysis for dsDNA 1000 Ladder
| Gel Lot # | Migration Time of 500 bp (s) | Peak Area %RSD (n=6 injections) | Resolution (500/517 bp) |
|---|---|---|---|
| A12345 | 15.2 ± 0.1 | 2.1% | 2.5 |
| B12346 | 14.8 ± 0.3 | 4.8% | 2.1 |
| A12347 | 15.1 ± 0.2 | 2.3% | 2.4 |
Table 3: Injection Parameter Influence on Peak Area Precision
| Injection Parameter | Value | %RSD of Peak Area (10 kDa Protein) | Sample Loading (nL, estimated) |
|---|---|---|---|
| Voltage (kV) | 5.0 | 3.5% | 4.2 |
| Time (s) | 10 | 2.8% | 3.8 |
| Voltage (kV) | 3.0 | 2.5% | 2.5 |
| Time (s) | 20 | 5.1% | 5.5 |
3. Detailed Experimental Protocols
Protocol 3.1: Assessing Capillary Coating Stability Objective: To monitor the performance degradation of a capillary coating over its operational lifetime. Materials: Coated capillary (e.g., hydrophilic polymer), CGE-LIF instrument, dsDNA or protein sizing ladder, run buffer, 1M NaOH, deionized water. Procedure:
- Condition new capillary with 1M NaOH (10 min), deionized water (5 min), and run buffer (10 min) at 5 psi.
- Perform 50 consecutive separations of the sizing ladder using standard method (e.g., -15 kV separation, 5 kV injection for 10 s).
- After every 10th run, introduce a control sample (e.g., a specific DNA fragment).
- Record migration time, peak efficiency (theoretical plates), and peak asymmetry for the target fragment in the control sample.
- Plot these parameters versus run number. A >10% shift in migration time or >20% loss in efficiency indicates coating failure.
Protocol 3.2: Qualifying New Gel Matrix Lots Objective: To validate the performance of a new gel matrix lot against established system suitability criteria. Materials: New and reference gel matrix lots, validated sizing ladder (e.g., dsDNA 1000), control sample (e.g., a monoclonal antibody). Procedure:
- Prepare separation matrix according to manufacturer instructions from new lot (Lot B) and a reference qualified lot (Lot A).
- Using the same capillary and instrument, perform six consecutive injections of the sizing ladder with Lot A, followed by six with Lot B.
- Calculate for key peaks (e.g., 100 bp, 500 bp): average migration time, %RSD of migration time, peak resolution, and peak area %RSD.
- Perform a statistical comparison (e.g., Student's t-test) of migration times between Lot A and Lot B. Significant difference (p < 0.05) indicates a need for method re-standardization.
- Document all data in a qualification report.
Protocol 3.3: Optimizing and Standardizing Electrokinetic Injection Objective: To establish a precise and robust electrokinetic injection protocol that minimizes bias. Materials: Standardized analyte at known concentration, plain run buffer, internal standard (if applicable). Procedure:
- Prepare a series of sample matrices with varying salt concentrations (0-50 mM NaCl) but constant analyte concentration.
- Using fixed voltage (e.g., 5 kV) and time (10 s), inject each sample in triplicate.
- Normalize peak areas to an internal standard if used. Plot normalized area vs. salt concentration. The plateau region indicates minimal matrix bias.
- Fix the sample matrix composition based on step 3. Then, perform a 2-factor DOE: Injection Voltage (2, 3, 5 kV) x Injection Time (5, 10, 15 s).
- Analyze a control sample in 6 replicates at the chosen optimal condition (typically lowest voltage/time giving sufficient signal and lowest %RSD).
4. Visualization of Workflows and Relationships
CGE-LIF Reproducibility Challenge Impact Map
CGE-LIF System Suitability Workflow
5. The Scientist's Toolkit: Essential Research Reagent Solutions
| Item | Function in CGE-LIF |
|---|---|
| Covalently Coated Capillaries (e.g., Hydrophilic Polymer) | Provides a stable, reproducible inner surface that minimizes electroosmotic flow (EOF) and analyte adsorption, critical for migration time precision. |
| Kit-Based Gel Matrix Buffers | Pre-mixed, viscosity-controlled polymer solutions (e.g., linear polyacrylamide) for sieving. Using kits from a single lot reduces run-to-run and inter-lab variability. |
| Fluorescent Intercalating Dye (e.g., SYBR Gold) | High-sensitivity dye for LIF detection of nucleic acids, allowing for precise peak area quantitation. Dye lot consistency is vital. |
| Universal Size Standards (dsDNA or Protein Ladder) | Used in every run to monitor capillary performance, gel integrity, and injection consistency. Acts as an internal system control. |
| Internal Standard (e.g., Fluorescently-labeled reference fragment) | Co-injected with the sample to normalize for injection volume variability, improving peak area precision. |
| Matrix-Consistent Sample Buffer | A low-conductivity buffer with matched ionic strength and additives (e.g., sucrose) to all samples, minimizing electrokinetic injection bias. |
Within the broader thesis on advancing Capillary Gel Electrophoresis with Laser-Induced Fluorescence detection (CGE-LIF) for biopharmaceutical analysis, a central challenge is the separation of complex, structurally similar analytes. This application note addresses the systematic optimization of three interdependent parameters—gel composition, buffer pH, and functional additives—to resolve challenging separations critical for drug development, such as oligonucleotide impurities, protein sizing, and glycan profiling.
Table 1: Optimization Matrix for Specific Analyte Classes
| Analyte Class | Challenge | Optimal Gel Type & Concentration | Critical pH Range | Key Additive(s) & Concentration | Impact on Resolution (Rs)* |
|---|---|---|---|---|---|
| siRNA / Oligonucleotides (20-25 bp) | Separation of N-1, N+1 impurities | Linear Polyacrylamide (LPA), 4-6% w/v | 8.0 - 9.0 (Tris-Borate-EDTA) | 7M Urea, 20-30% Formamide | Rs increases from <1.0 to >2.5 for N-1 peak |
| Protein Sizing (10-225 kDa) | Resolution of aggregates & fragments | Coated Capillary, LPA 3-5% w/v | 8.0 - 8.8 (Tris-Glycine) | 0.1% SDS (critical), 10% Glycerol | Enables baseline resolution of monomer/aggregate (Rs >1.5) |
| Released N-Glycans (APTS-labeled) | Isomer separation (α2-3 vs α2-6 sialylation) | High-Density LPA, 8-10% w/v | 4.5 - 5.0 (Ammonium Acetate) | 10-15 mM γ-Cyclodextrin | Resolves sialic acid linkage isomers (ΔMigration >0.5 min) |
| Antisense Oligonucleotides (ASOs) | Phosphorothioate (PS) diastereomer separation | High-Performance LPA, 10% T | 8.5 - 9.5 | 25-50 mM Magnesium Chloride (MgCl₂) | Partial resolution of PS diastereomers observed |
Rs (Resolution) calculated as 2(t₂ - t₁)/(w₁ + w₂), where t is migration time and w is peak width.
Table 2: Effect of Additives on Separation Performance Metrics
| Additive | Primary Function | Optimal Concentration Range | Key Parameter Affected | Direction of Change | Consideration |
|---|---|---|---|---|---|
| Urea | Denaturant, disrupts secondary structure | 4 - 8 M | Migration time reproducibility | Improves (CV < 1%) | High concentrations increase current/viscosity. |
| Glycerol | Viscosity modifier, reduces EOF | 5 - 15% v/v | Run-to-run precision | Improves | High % increases run time. |
| Cyclodextrins (γ) | Host-guest interactions, chiral separation | 5 - 20 mM | Selectivity (α) | Modulates | Type and conc. are analyte-specific. |
| MgCl₂ / Divalent Cations | Charge masking, conformation control | 10 - 50 mM | Peak shape for polyanions | Sharpens | Can cause precipitation; requires screening. |
| Formamide | Denaturant, lowers melting temperature | 15 - 30% v/v | Resolution of small fragments | Increases | Can be combined with urea. |
Experimental Protocols
Protocol 1: Optimized CGE-LIF for siRNA Impurity Analysis
- Objective: Baseline separation of a 21-mer siRNA from its N-1 (20-mer) and N+1 (22-mer) synthesis impurities.
- Materials: See "The Scientist's Toolkit" (Section 5).
- Method:
- Capillary: 50 µm i.d., 30 cm effective length (40 cm total), bare fused silica.
- Gel Matrix Preparation: Prepare a 5% w/v Linear Polyacrylamide (LPA) sieving matrix in 1x TBE buffer (pH 8.3). Dissolve urea to a final concentration of 7M. Filter through a 0.45 µm syringe filter.
- Sample Prep: Dilute siRNA sample to 0.1 mg/mL in nuclease-free water containing 1 mM EDTA.
- Instrument Conditions:
- Detection: LIF (Ex/Em: 488/520 nm).
- Pre-run: 5 min at 300 V/cm.
- Injection: Electrokinetic, 5 kV for 10 s.
- Separation Voltage: 300 V/cm (constant).
- Temperature: 30°C.
- Optimization Step: If resolution is insufficient, incrementally increase formamide concentration in the gel matrix from 0% to 25% (v/v) and re-evaluate.
Protocol 2: High-Resolution Glycan Isomer Separation
- Objective: Separate APTS-labeled biantennary glycans differing in sialic acid linkage (α2-3 vs α2-6).
- Materials: See "The Scientist's Toolkit" (Section 5).
- Method:
- Capillary: 50 µm i.d., 30 cm effective length, covalently coated for EOF suppression.
- Gel/Buffer Preparation: Prepare a high-density 9% w/v LPA gel in 50 mM ammonium acetate buffer, pH 4.75. Add γ-Cyclodextrin to a final concentration of 12 mM. Sonicate to dissolve.
- Sample Prep: Use commercially APTS-labeled glycans. Dilute in water.
- Instrument Conditions:
- Detection: LIF (Ex/Em: 488/520 nm).
- Pre-run: 3 min at 200 V/cm.
- Injection: Pressure, 0.5 psi for 10 s.
- Separation Voltage: 400 V/cm (constant).
- Temperature: 25°C.
- Optimization Step: Fine-tune selectivity by adjusting pH in 0.1-unit increments between 4.5 and 5.0, and γ-cyclodextrin concentration between 5-15 mM.
Visualization Diagrams
Diagram Title: CGE-LIF Method Optimization Workflow
Diagram Title: CGE Separation Parameter Interplay
The Scientist's Toolkit: Research Reagent Solutions
| Item / Reagent | Function in CGE-LIF Optimization | Example Product/Chemical |
|---|---|---|
| Linear Polyacrylamide (LPA) | Non-cross-linked sieving polymer; concentration dictates pore size and resolution range. | Ready-to-use LPA gels or acrylamide/bis-acrylamide for in-lab polymerization. |
| Tris-Borate-EDTA (TBE) Buffer | Standard conductive buffer for nucleic acids; pH ~8.3 maintains deprotonated phosphate backbone. | Molecular biology grade 10x TBE concentrate. |
| Urea (High-Purity) | Denaturant; disrupts secondary structure in nucleic acids and proteins, ensuring separation by size. | Ultrapure grade, deionized for stability. |
| γ-Cyclodextrin | Chiral additive; interacts with glycan isomers or stereoisomers to impart differential mobility. | Pharmaceutical secondary standard. |
| SDS (Sodium Dodecyl Sulfate) | Anionic detergent; binds proteins uniformly by mass, enabling size-based separation (CE-SDS). | Electrophoresis purity grade. |
| Formamide | Denaturant; lowers nucleic acid melting temperature (Tm), improving resolution of fragments. | Molecular biology grade, deionized. |
| Coated Capillaries | Suppress/eliminate electroosmotic flow (EOF) and analyte adsorption to the capillary wall. | e.g., Polyvinyl alcohol (PVA) coated capillaries. |
| APTS (8-Aminopyrene-1,3,6-Trisulfonate) | Fluorescent dye for labeling glycans; provides high sensitivity via LIF detection. | >95% purity, for glycan labeling kits. |
Preventative Maintenance and Best Practices for System Longevity and Data Quality
1. Introduction Within a CGE-LIF research framework, system reliability and data integrity are paramount. Preventative maintenance (PM) is not merely operational but a critical scientific control. This document outlines application notes and protocols to ensure capillary electrophoresis (CE) system longevity and uphold the high data quality required for sensitive applications like biopharmaceutical analysis and glycan profiling.
2. Preventative Maintenance Schedule and Procedures Adherence to a strict PM schedule minimizes downtime and variability. Key tasks are summarized below.
Table 1: Recommended Preventative Maintenance Schedule for CGE-LIF Systems
| Component | Frequency | Action | Purpose |
|---|---|---|---|
| Capillary | Pre/Post-run | Flush with 0.1M NaOH, ddH₂O, and run buffer. | Remove adsorbed species, prevent clogging. |
| Every 50 runs | Perform a more aggressive wash (e.g., 1M HCl, 0.1M NaOH). | Degrade persistent contaminants. | |
| Autosampler | Weekly | Clean sample tray and probe with ddH₂O/isopropanol. | Prevent cross-contamination and salt buildup. |
| Laser & Optics | Monthly | Inspect for dust; clean external windows with optical-grade solvent/lens paper. | Maintain optimal excitation intensity and detection sensitivity. |
| Buffer Vials | Each run | Replace with fresh, filtered (0.22 µm) buffer. | Prevent microbial growth and particulate introduction. |
| Data Backup | Daily/Weekly | Automated backup to secure, off-instrument storage. | Ensure data integrity and traceability. |
3. Protocol for Critical Performance Qualification (PQ) Experiment Regular PQ verifies system suitability against predefined performance criteria.
Title: Quarterly Performance Qualification of CGE-LIF System Using a Standardized Ladder Objective: To assess resolution, migration time reproducibility, and signal-to-noise ratio (S/N). Materials:
- CGE-LIF System (e.g., PA 800 Plus or equivalent)
- N-CHO coated capillary (e.g., 50 µm i.d., 30 cm total length)
- Gel Matrix: Commercial carbohydrate or protein separation gel.
- Standard: 480 Ladder (for glycosylation analysis) or 10/100 kDa protein ladder.
- Running Buffer: As specified for the gel matrix.
Procedure:
- System Preparation: Install a new or freshly reconditioned capillary. Prime the system with gel matrix for 20 minutes at high pressure (e.g., 100 psi).
- Sample Preparation: Dilute the standard ladder per manufacturer's instructions in ddH₂O.
- Injection: Electrokinetically inject sample at 1-5 kV for 5-10 seconds.
- Separation: Run at constant voltage (e.g., 15-20 kV) for 30 minutes. Maintain capillary temperature at 20-25°C.
- Detection: Use LIF with appropriate excitation/emission filters (e.g., 488 nm/520 nm).
- Replicates: Perform six consecutive injections from the same sample vial.
- Data Analysis: Calculate the following from the electropherograms:
- Migration Time RSD% for a key peak.
- Resolution (Rs) between two closely migrating peaks: Rs = 2(t₂ - t₁) / (w₁ + w₂).
- Signal-to-Noise Ratio (S/N) for the smallest peak.
Acceptance Criteria: Migration Time RSD < 1.0%; Rs > 1.5; S/N > 10:1. Failure mandates investigation and corrective action.
4. The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagent Solutions for CGE-LIF Maintenance and Operation
| Reagent/Material | Function & Importance |
|---|---|
| 0.1M & 1.0M Sodium Hydroxide (NaOH) | Primary capillary wash solution. Hydrolyzes adsorbed proteins and contaminants. |
| 0.1M & 1.0M Hydrochloric Acid (HCl) | Alternative/sequential wash for removing cationic contaminants and rinsing NaOH. |
| Deionized Water (18.2 MΩ·cm) | Universal rinse solvent to remove salts and acids/bases before introducing buffer/gel. |
| CE-Grade Separation Gel/Matrix | Contains sieving polymer (e.g., linear polyacrylamide). Critical for size-based resolution. Must be filtered and degassed. |
| Fluorescent Derivatization Dye (e.g., 8-AP, ANTS) | Tags non-fluorescent analytes (glycans, amines) for LIF detection. Purity is critical for low background. |
| Validated System Suitability Standard | Certified ladder (e.g., NIST mAb) to track system performance over time and across labs. |
5. CGE-LIF System Health Monitoring & Troubleshooting Workflow A logical decision pathway ensures consistent data quality.
Diagram 1: CGE-LIF System Health Check Workflow (91 chars)
6. Signaling Pathway for Data Degradation and Countermeasures This diagram outlines root causes of poor data quality and corresponding preventative actions.
Diagram 2: Data Quality Failure Modes and Preventative Actions (88 chars)
CGE-LIF vs. Other Techniques: Validation Strategies, Comparability, and Regulatory Considerations
Within the context of a broader thesis on Capillary Gel Electrophoresis with Laser-Induced Fluorescence detection (CGE-LIF) for biopharmaceutical analysis, establishing a rigorous validation framework is paramount. This framework ensures the method is fit for its intended purpose, such as monitoring critical quality attributes (CQAs) like size heterogeneity, glycosylation patterns, or aggregation of monoclonal antibodies and gene therapies. This document provides detailed application notes and protocols for validating key analytical performance characteristics.
Experimental Protocols & Data
Protocol: Assessment of Specificity and Selectivity
Objective: To demonstrate the method's ability to distinguish the target analyte from other components in the sample matrix (e.g., excipients, degradants, process-related impurities). Materials: Sample Buffer (e.g., 50 mM Tris-Borate-EDTA, pH 8.3), Sieving Gel Matrix (e.g., dextran- or polyacrylamide-based), Fluorescent Intercalating Dye (e.g., SYBR Gold), Bare Fused-Silica Capillary (50 µm i.d., total length 30 cm, effective length 20 cm). Procedure:
- Prepare sample set: a) Analyte standard (e.g., 20 kDa ssDNA ladder). b) Matrix blank (formulation buffer). c) Stressed analyte (heat-induced aggregates). d) Spiked matrix (analyte in formulation buffer).
- Inject samples hydrodynamically (0.5 psi for 10 s).
- Perform electrophoresis at -10 kV for 15 minutes with gel matrix filling the capillary.
- Use LIF detection (excitation: 488 nm, emission: 520 nm).
- Analyze electropherograms for resolution of peaks and absence of matrix interference at the analyte migration time. Acceptance Criteria: The analyte peak is baseline resolved (R_s > 1.5) from all other peaks. No significant peaks co-migrate with the analyte in the blank.
Protocol: Assessment of Linearity and Range
Objective: To evaluate the proportionality of the detector response across a defined concentration range of the analyte. Procedure:
- Prepare a minimum of five standard solutions covering 50-150% of the expected sample concentration range (e.g., 0.5-50 µg/mL for a protein).
- Analyze each standard in triplicate in randomized order.
- Plot peak area (or corrected peak area) vs. concentration.
- Perform least-squares linear regression analysis. Acceptance Criteria: Correlation coefficient (r) ≥ 0.990. Residuals are randomly scattered.
Table 1: Linearity Data for a 10-100 µg/mL Monoclonal Antibody Fragment
| Concentration (µg/mL) | Mean Peak Area (n=3) | Standard Deviation |
|---|---|---|
| 10 | 12540 | 450 |
| 25 | 29875 | 780 |
| 50 | 60550 | 1120 |
| 75 | 92010 | 1850 |
| 100 | 122500 | 2500 |
| *Regression Equation: y = 1225.5x - 550.2 | r² = 0.9987* |
Protocol: Assessment of Precision (Repeatability & Intermediate Precision)
Objective: To measure the closeness of agreement between a series of measurements under prescribed conditions. Procedure for Repeatability (Intra-day):
- Prepare six independent sample preparations at 100% of the test concentration.
- Analyze all six sequentially by one analyst on one instrument in one day.
- Calculate %RSD for migration time and peak area. Procedure for Intermediate Precision:
- Repeat the repeatability study on a different day, with a different analyst, and/or on a different instrument (per a predefined experimental design).
- Use analysis of variance (ANOVA) to assess the significance of the between-day/analyst/instrument variances. Acceptance Criteria: %RSD for migration time ≤ 1.0%; %RSD for peak area ≤ 5.0% for repeatability. No significant difference (p > 0.05) between conditions for intermediate precision.
Protocol: Determination of LOD and LOQ
Objective: To establish the lowest concentration of analyte that can be reliably detected (LOD) and quantified (LOQ). Procedure (Signal-to-Noise Method):
- Prepare a series of low-concentration standards near the expected detection limit.
- Inject and analyze each standard.
- Measure the signal-to-noise ratio (S/N) by comparing the analyte peak height (H) to the baseline noise (N), where N is the peak-to-peak noise over a defined region.
- LOD: Concentration yielding S/N ≈ 3.
- LOQ: Concentration yielding S/N ≈ 10. Procedure (Standard Deviation of Response and Slope):
- Determine the standard deviation (σ) of the y-intercept of the regression line or the standard deviation of the response for low-concentration samples.
- Calculate: LOD = 3.3σ / S; LOQ = 10σ / S, where S is the slope of the calibration curve.
Table 2: LOD/LOQ Determination for ssDNA Impurity
| Method | LOD (ng/mL) | LOQ (ng/mL) |
|---|---|---|
| Signal-to-Noise (S/N) | 2.1 | 6.5 |
| SD of Response/Slope | 1.8 | 5.4 |
Protocol: Assessment of Robustness
Objective: To measure the method's capacity to remain unaffected by small, deliberate variations in operational parameters. Procedure (via Experimental Design):
- Identify critical method parameters: e.g., Run Temperature (± 2°C), Buffer pH (± 0.2 units), Gel Matrix Lot, Capillary Batch.
- Design a Plackett-Burman or fractional factorial experiment to evaluate these factors at two levels.
- Analyze a system suitability sample (e.g., a defined mixture) under each experimental condition.
- Monitor Critical Responses: Migration time of main peak, resolution between two critical peaks, peak area.
- Use statistical analysis (e.g., ANOVA, Pareto charts) to identify significant effects.
Table 3: Robustness Test Results - Effects on Main Peak Migration Time
| Parameter | Variation Level | Mean Migration Time (min) | %RSD | Significant (p < 0.05)? |
|---|---|---|---|---|
| Temperature | +2°C | 8.45 | 0.6 | No |
| -2°C | 8.62 | |||
| Buffer pH | +0.2 | 8.50 | 1.1 | No |
| -0.2 | 8.58 | |||
| Gel Lot | Lot A | 8.52 | 2.5 | Yes |
| Lot B | 8.73 |
The Scientist's Toolkit: Research Reagent Solutions
| Item/Reagent | Function in CGE-LIF |
|---|---|
| Dextran/Polyacrylamide Gel Matrix | Acts as a molecular sieve, separating analytes (e.g., proteins, nucleic acids) based on size. |
| Fluorescent Intercalating Dye (e.g., SYBR Gold, POPO-3) | Non-covalently binds to nucleic acids or proteins, enabling high-sensitivity LIF detection. |
| Coated Capillary (e.g., DB-1) | Reduces analyte-wall interactions (adsorption), improving peak shape and reproducibility for proteins. |
| Size Standards (e.g., DNA/RNA Ladder, Protein Ladder) | Used for system suitability and to construct a calibration curve for size determination. |
| Sample/Stacking Buffer (Low Ionic Strength) | Promotes sample stacking at the capillary inlet, enhancing detection sensitivity. |
| Run Buffer (e.g., TBE with SDS) | Provides consistent ionic strength and pH for stable electrophoresis; SDS denatures proteins for size-based separation. |
Diagrams
Title: CGE-LIF Method Validation Workflow
Title: CGE-LIF Detection Principle
Title: CGE-LIF Robustness Test Parameters
Within the broader thesis on Capillary Gel Electrophoresis with Laser-Induced Fluorescence detection (CGE-LIF), this analysis contextualizes its role among predominant techniques for protein purity, size, and aggregation analysis in biopharmaceutical development. CGE-LIF, offering high sensitivity and quantitative precision, is positioned against traditional SDS-PAGE, mainstream CE-SDS (UV), and orthogonal SEC-MALS. This document provides a detailed comparative framework, application notes, and protocols to guide researchers in method selection.
Table 1: Comparative Overview of Techniques
| Feature | SDS-PAGE (Traditional) | CE-SDS (UV) | SEC-MALS | CGE-LIF (Thesis Context) |
|---|---|---|---|---|
| Principle | Size-based migration in polyacrylamide gel under electric field. | Size-based migration in a sieving polymer-filled capillary. | Size-based separation by HPLC coupled to multi-angle light scattering. | Size-based migration in a sieving polymer-filled capillary with LIF detection. |
| Detection Mode | Coomassie/ silver stain, fluorescence imaging. | UV absorbance at 214/220 nm. | Refractive Index (RI), UV, Light Scattering (LS). | Laser-Induced Fluorescence (high sensitivity). |
| Sample Throughput | Low (manual, batch). | Medium-High (automated). | Low-Medium (serial HPLC runs). | High (automated, multi-capillary systems possible). |
| Quantitation | Semi-quantitative (densitometry). | Quantitative (peak area). | Quantitative (absolute mass). | Highly quantitative, superior linear dynamic range. |
| Resolution | Moderate. | High. | Moderate (based on column). | Very High (efficient capillary-based separation). |
| Mass Accuracy | Low (relative to ladder). | Medium (relative to ladder). | High (absolute, from first principles). | Medium (relative to ladder). |
| Aggregation Detection | Yes, but poor resolution of low levels. | Yes, good for non-covalent aggregates under denaturing conditions. | Excellent (direct, native or denaturing). | Yes, excellent sensitivity for low-abundance species. |
| Sample Consumption | ~5-20 µg. | ~1-10 ng (UV). | ~10-100 µg. | <1 ng (attomole sensitivity with labeling). |
| Key Strength | Low cost, widespread use, visual result. | Automated, quantitative, good for purity charge variant orthogonal analysis. | Absolute molecular weight without standards, native state analysis. | Ultra-sensitive, ideal for low-concentration samples (e.g., CQAs from cell lysates). |
| Key Weakness | Poor quantitation, low throughput, labor-intensive. | Lower sensitivity than LIF, may miss low-level impurities. | Lower resolution than CE, higher sample need, method development complexity. | Requires fluorescent dye labeling (additional step), not absolute mass. |
Table 2: Typical Performance Metrics (Representative Data)
| Metric | SDS-PAGE | CE-SDS (UV) | SEC-MALS | CGE-LIF |
|---|---|---|---|---|
| Limit of Detection (LOD) | ~1-10 ng/band | ~0.1 mg/mL (injection conc.) | ~0.01 mg/mL | ~0.001 mg/mL (or pg/band) |
| Run Time | 60-90 min (+ staining) | 20-45 min | 15-30 min | 20-45 min |
| %RSD (Migration Time) | >5% | 0.5-2% | 0.5-2% (retention time) | 0.3-1.5% |
| %RSD (Peak Area) | 10-25% | 1-5% | 2-5% | 1-3% |
| Linear Dynamic Range | ~1 order of magnitude | 2-3 orders of magnitude | 2-3 orders of magnitude | 3-4 orders of magnitude |
Detailed Protocols
Protocol 1: Reduced CE-SDS (UV) for Monoclonal Antibody Purity Analysis
Application Note: Standard purity assay for release and stability testing of mAbs.
- Sample Preparation: Dilute antibody to 2 mg/mL in PBS. For reduced analysis, mix 50 µL sample with 10 µL 0.5M iodoacetamide (alkylation) and 25 µL 10% w/v SDS. Incubate at 70°C for 5 min. Add 25 µL of 10% β-mercaptoethanol, incubate at 70°C for 5 min.
- Instrument Setup: Use a CE system (e.g., Beckman Coulter PA 800 Plus) with a bare-fused silica capillary (50 µm i.d., 30.2 cm total length, 20 cm effective length). Fill capillary with SDS-MW gel solution.
- Separation Parameters: Inject sample electrokinetically at 5 kV for 20 sec. Separate at constant voltage of 15 kV for 30 min, with cartridge temperature at 25°C. Detect at 220 nm.
- Data Analysis: Integrate peaks for light chain (LC) and heavy chain (HC). Calculate % purity as (Area LC + Area HC) / Total area of all peaks × 100. Compare migration times to a pre-stained protein ladder.
Protocol 2: SEC-MALS for Absolute Molecular Weight and Aggregation
Application Note: Determine absolute molecular weight and quantify aggregates under native conditions.
- System Equilibration: Use an HPLC system coupled to a MALS detector (e.g., Wyatt Dawn Heleos II) and RI detector (e.g., Wyatt Optilab T-rEX). Equilibrate a size-exclusion column (e.g., Tosoh TSKgel G3000SWxl) with mobile phase (0.1M sodium phosphate, 0.1M sodium sulfate, pH 6.7) at 0.5 mL/min until stable baseline.
- Sample Preparation & Injection: Centrifuge protein sample at 14,000g for 10 min. Filter through 0.1 µm spin filter. Inject 50 µL of sample at 1 mg/mL.
- Data Collection: Run isocratic elution for 30 min. Collect light scattering data at multiple angles and RI signal simultaneously.
- Analysis (Astra Software): Determine the differential refractive index increment (dn/dc) value (typically 0.185 mL/g for proteins). Process data using the Zimm or Debye model to calculate absolute molecular weight across the elution peak. Integrate monomer and aggregate peak areas from the RI chromatogram for % aggregation.
Protocol 3: CGE-LIF for High-Sensitivity Glycoprotein Heterogeneity
Application Note (Thesis Context): Profile charge/size variants of low-abundance glycoproteins from limited sample sources (e.g., cell culture supernatants).
- Fluorescent Labeling: Use a fluorophore like Chromeo P503. Mix 5 µL of protein sample (0.1-0.5 mg/mL) with 2 µL of 1M borate buffer (pH 9.0) and 3 µL of Chromeo P503 dye (1 mM in DMSO). Incubate at room temperature in the dark for 30 min.
- Quenching & Dilution: Add 5 µL of 1M Tris-HCl (pH 7.0) to quench the reaction. Dilute the mixture 1:10 with deionized water.
- CGE-LIF Analysis: Use a CGE-LIF system (e.g., Beckman Coulter ProteomeLab PA 800 with LIF). Capillary: 50 µm i.d., 30 cm total length (20 cm to detector). Gel matrix: Linear polyacrylamide or PEG-based sieving matrix.
- Run Conditions: Inject sample at 5 kV for 10 sec. Separate at 15 kV constant voltage, 25°C. LIF detection: Excitation 488 nm, Emission 520 nm.
- Data Processing: Analyze electrophoregram peaks for main species and micro-heterogeneity (e.g., glycoforms). Use an internal fluorescent standard for precise migration time normalization. Quantify variant percentages based on peak area.
Visualizations
Technique Selection Decision Tree
CGE-LIF Experimental Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for CGE-LIF and Comparative Techniques
| Item | Function/Application | Example Vendor/Product |
|---|---|---|
| Fluorescent Dye (Amino-reactive) | Covalently labels proteins for LIF detection. Critical for CGE-LIF sensitivity. | Chromeo P503 (Active Motif), Alexa Fluor 488 NHS ester (Thermo Fisher). |
| SDS-MW Gel Solution | Sieving matrix for CE-SDS (UV & LIF) providing size-based separation. | Beckman Coulter SDS-MW Gel Buffer, Bio-Rad CE-SDS Run Buffer. |
| Reducing & Alkylating Agents | Denature and break disulfide bonds for reduced analysis (SDS-PAGE, CE-SDS). | β-Mercaptoethanol, DTT, Iodoacetamide. |
| Pre-stained Protein Ladder | Provides molecular weight calibration in SDS-PAGE and CE-SDS. | Precision Plus Protein Standard (Bio-Rad), HiMark Standard (Thermo). |
| MALS-Compatible SEC Columns | HPLC columns for native size separation without protein interaction for SEC-MALS. | TSKgel from Tosoh Bioscience, AdvanceBio SEC from Agilent. |
| MALS & RI Detectors | Measure light scattering and concentration for absolute molecular weight. | DAWN (MALS) & Optilab (RI) from Wyatt Technology. |
| Bare Fused Silica Capillaries | The separation channel for CE-based methods (CE-SDS, CGE-LIF). | Beckman Coulter, Polymicro Technologies. |
| High-Purity SDS | Critical for consistent protein-SDS complex formation in SDS-based methods. | Sodium Dodecyl Sulfate, >99% purity (e.g., MilliporeSigma). |
| dn/dc Value Database/Standard | Reference for protein refractive index increment in MALS calculations. | BSA standard for validation, literature values. |
| Micro-Sample Vials & Caps | For low-volume, high-precision sample handling in automated CE and HPLC. | 0.2 mL PCR tubes or specific instrument vials (e.g., Agilent). |
1. Introduction Within CGE-LIF research for biopharmaceutical analysis, maintaining data comparability across the product lifecycle—from early development to quality control (QC)—is paramount. This document outlines application notes and protocols to bridge method parameters, ensuring consistent, reliable characterization of critical quality attributes (CQAs) like protein size heterogeneity, glycan profiling, and oligonucleotide purity.
2. Application Notes: Key Parameters for Lifecycle Bridging Successful method transfer hinges on controlling specific parameters. The table below summarizes target performance criteria and typical acceptance limits for a CGE-LIF method analyzing a monoclonal antibody size heterogeneity assay.
Table 1: CGE-LIF Method Performance Criteria for Size Variant Analysis
| Parameter | Development Phase Target | QC Phase Acceptance Criterion | Justification |
|---|---|---|---|
| Migration Time RSD | < 1.0% (n=10) | ≤ 2.0% (n=6) | Indicates run-to-run capillary and instrumental stability. |
| Peak Area RSD (Main Peak) | < 1.5% (n=10) | ≤ 3.0% (n=6) | Ensures quantitative reproducibility of the dominant species. |
| Size Variant Relative % RSD | < 5.0% (n=10) | ≤ 10.0% (n=6) | Controls precision for low-abundance variants (e.g., aggregates, fragments). |
| Resolution (Main Peak/Aggregate) | ≥ 2.0 | ≥ 1.8 | Maintains ability to discriminate critical species. |
| LOD (for fragment) | ≤ 0.1% | Not Applicable | Development phase sensitivity requirement. |
| System Suitability | Daily check with reference standard | Each analysis batch | Ensures the system is performing as validated. |
3. Experimental Protocols
Protocol 3.1: Standardized CGE-LIF for Protein Size Heterogeneity Objective: To separate and quantify high-molecular-weight (HMW) aggregates, main monomer, and low-molecular-weight (LMW) fragments of a monoclonal antibody.
Materials:
- Gel-filled capillary (e.g., 50 µm i.d., 30 cm total length, 20 cm effective length).
- CGE-LIF instrument with 488 nm excitation laser.
- Proprietary sieving gel buffer (e.g., containing SDS).
- Fluorescent dye for non-covalent labeling (e.g., intercalating dye).
- Sample buffer (commercial, containing SDS and internal standard).
- Reference standard and system suitability sample.
- Deionized water.
Procedure:
- Instrument Setup: Install capillary cartridge. Set detector to appropriate gain/PMT voltage. Set temperature to 25°C.
- Conditioning: Pre-run conditioning with fresh gel buffer for 5 min at 500 V/cm.
- Sample Preparation: Dilute protein sample to 1 mg/mL in sample buffer. Add fluorescent dye per manufacturer's instructions. Vortex and incubate at 65°C for 5 min. Centrifuge briefly.
- Injection: Hydrodynamic injection (e.g., 3.0 kPa for 30 sec).
- Separation: Apply constant voltage of 500 V/cm for 20 minutes.
- Data Analysis: Integrate peaks. Identify species via migration time relative to internal standard. Calculate % area for each peak relative to total integrated area.
Protocol 3.2: Method Transfer and Comparability Study Objective: To validate method performance in a QC lab against development lab data.
Procedure:
- Pre-Transfer: Document all method parameters, instructions, and acceptance criteria (Table 1). Ship identical reagents, capillaries, and reference standards.
- Training: QC analysts perform 3 runs under supervision of development scientist.
- System Suitability: QC lab performs 6 consecutive runs of the system suitability sample. All results must meet pre-defined QC acceptance criteria.
- Sample Analysis: Analyze 3 lots of product in triplicate. Calculate mean and RSD for all CQAs.
- Statistical Comparison: Perform equivalence testing (e.g., two one-sided t-tests) between development and QC lab results for each CQA. The 90% confidence interval for the difference must fall within ±1.5%.
4. Visualizations
Diagram Title: Method Lifecycle from Development to QC
Diagram Title: CGE-LIF Analytical Workflow
5. The Scientist's Toolkit
Table 2: Key Research Reagent Solutions for CGE-LIF
| Item | Function in CGE-LIF |
|---|---|
| Fluorescent Non-Covalent Dye (e.g., intercalator) | Binds non-covalently to SDS-protein complexes, enabling highly sensitive LIF detection without pre-column labeling artifacts. |
| Proprietary SDS-MW Gel Buffer | Provides a dynamic sieving matrix for size-based separation; lot-to-lot consistency is critical for comparability. |
| Carboxylated Coated Capillary | Minimizes electroosmotic flow (EOF) and protein adsorption, ensuring reproducible migration. |
| Internal Standard (fluorescently labeled) | A consistent, stable compound co-injected to normalize migration time and correct for injection variability. |
| System Suitability Reference Standard | A well-characterized control sample to verify instrument and method performance meets specifications before sample analysis. |
| Sample Buffer with Reducing Agent | Denatures and linearizes proteins with SDS; DTT or similar agent breaks disulfide bonds for accurate size analysis. |
The development and quality control of biologics, including monoclonal antibodies (mAbs), antibody-drug conjugates (ADCs), and gene therapies, require stringent analytical method validation and lifecycle management. The International Council for Harmonisation (ICH) guidelines Q2(R2) on analytical procedure validation and the newly adopted Q14 on analytical procedure development provide the modern framework. These are complemented by specific FDA and EMA guidelines for biologics (e.g., FDA's "Analytical Procedures and Methods Validation for Drugs and Biologics," EMA's "Guideline on development, production, characterisation and specifications for monoclonal antibodies and related products").
Within this regulatory context, Capillary Gel Electrophoresis with Laser-Induced Fluorescent detection (CGE-LIF) has emerged as a critical, high-resolution technique for the analysis of size variants (e.g., fragments and aggregates) and charge variants of proteins, as well as for the purity and size analysis of oligonucleotides. Its superior sensitivity, quantitation, and reproducibility align directly with regulatory demands for robust, validated methods.
Application Notes: Aligning CGE-LIF Applications with Regulatory Guidelines
Purity and Impurity Analysis of mAbs (ICH Q2(R2) / Q5E)
CGE-LIF, using SDS-MW separation, is a validated alternative to traditional SDS-PAGE for assessing protein purity, fragments, and aggregates. Regulatory guidelines emphasize the need for methods that can detect and quantify product-related impurities.
Key Quantitative Data Summary: Table 1: Method Validation Parameters for mAb Purity Assay by CGE-LIF (SDS-MW Kit)
| Validation Parameter | ICH Q2(R2) Requirement | Typical CGE-LIF Performance | Acceptance Criteria (Example) |
|---|---|---|---|
| Specificity | Unambiguous detection of analyte | Baseline separation of mAb monomer from fragment and aggregate peaks. | No interference from buffer/excipient blanks. |
| Linearity & Range | Directly proportional relationship | R² > 0.99 for monomer from 0.1 to 2.0 mg/mL. | R² ≥ 0.990 |
| Precision (Repeatability) | Closeness of agreement under same conditions | %RSD of migration time < 1.0%; %RSD of peak area < 5.0%. | %RSD ≤ 2.0% (time), ≤ 10.0% (area) |
| Intermediate Precision | Variation within/labs, analysts, days | Combined %RSD of peak area < 7.0% across multiple runs. | %RSD ≤ 15.0% |
| Accuracy / Recovery | Closeness to true value | Spike recovery of known fragments: 95-105%. | 80-120% |
| Detection Limit (LOD) | Lowest detectable amount | ~0.1% of main peak (with LIF). | Visual evaluation or S/N ≥ 3 |
| Quantitation Limit (LOQ) | Lowest quantifiable amount | ~0.5% of main peak (with LIF). | %RSD ≤ 20%, S/N ≥ 10 |
| Robustness | Insensitivity to deliberate variations | Evaluated for buffer age, voltage, temperature variations. | System suitability criteria met. |
Critical Quality Attribute (CQA) Monitoring for ADCs (ICH Q14 / Q8)
ICH Q14 encourages analytical procedure design based on risk assessment and understanding. For ADCs, the Drug-to-Antibody Ratio (DAR) distribution and unconjugated antibody are critical CQAs. CGE-LIF in a non-reduced, native-like conditions can separate and quantify species with different DARs.
Table 2: Application of CGE-LIF for ADC Characterization
| ADC Attribute | Relevant Guideline | CGE-LIF Role | Measurable Output |
|---|---|---|---|
| DAR Distribution | ICH Q8(R2), Q14 | Separation of DAR species (DAR0, DAR2, DAR4, etc.) based on hydrodynamic size/charge shift. | Relative percentage of each DAR species. |
| Unconjugated mAb (DAR0) | FDA Guidance for Industry: ADC | Sensitive quantification of free antibody. | % DAR0 (Target: < 5-10%) |
| High Molecular Weight Species | ICH Q5C, Q6B | Detection of aggregates induced by conjugation process. | % Aggregate (Target: < 2-5%) |
Oligonucleotide Purity and Identity (Relevant to mRNA/vaccines)
For synthetic oligonucleotides and mRNA-based therapeutics, CGE-LIF is the gold standard for assessing identity, purity, and detecting short/longmers. ICH Q6B and relevant FDA guidance are applicable.
Table 3: CGE-LIF Validation for Oligonucleotide Analysis
| Parameter | Typical Specification | CGE-LIF Method Outcome |
|---|---|---|
| Identity (Size) | Conformance to expected length | Migration time correlated to DNA/RNA ladder. |
| Purity (Full-length) | ≥ 80% full-length product | Quantitation of main peak vs. failure sequences. |
| Resolution (N vs. N-1) | Baseline separation | Resolution factor ≥ 1.5. |
| LOQ for Impurities | Report, identify, control thresholds | Typically < 0.1% relative abundance. |
Experimental Protocols
Protocol 1: Validation of a CGE-LIF Method for mAb Purity and Aggregate Analysis
Title: Validation of Purity and Aggregate Assay for Therapeutic mAb using CGE-LIF (SDS-MW Kit).
Objective: To perform a partial validation (per ICH Q2(R2)) of a CGE-LIF method for the quantification of monomer, fragment, and aggregate content of a therapeutic monoclonal antibody.
Materials: See "The Scientist's Toolkit" below.
Methodology:
- Sample Preparation:
- Dilute the mAb reference standard and test samples to 1 mg/mL using deionized water.
- Prepare the sample buffer: Mix 45 μL of SDS-MW sample buffer with 5 μL of 2-mercaptoethanol (for reduced analysis) or 5 μL of deionized water (for non-reduced analysis) per sample.
- Combine 25 μL of diluted sample with 25 μL of the prepared sample buffer.
- Heat the mixture at 70°C for 5 minutes. Cool to room temperature before injection.
Instrument Setup (CGE-LIF):
- Instrument: PA 800 Plus or similar.
- Detection: LIF with 488 nm excitation / 520 nm emission filter.
- Capillary: 50 μm ID, 30.2 cm total length (20 cm effective) bare fused silica.
- Gel Matrix: Commercial SDS-MW gel matrix.
- Run Buffer: Commercial SDS-MW run buffer.
- Conditions: -15 kV (reverse polarity) for 30 minutes. Temperature: 25°C.
- Injection: Electrokinetic injection at -5 kV for 20 seconds.
Validation Experiments:
- Specificity: Analyze blank buffer, placebo formulation, and a stressed mAb sample (heat-induced aggregation/ fragmentation).
- Linearity & Range: Prepare a series of mAb standard solutions from 0.1 mg/mL to 2.0 mg/mL (n=5 concentrations, triplicate injections). Plot peak area vs. concentration.
- Precision (Repeatability): Inject six independently prepared samples from a homogeneous mAb lot (1 mg/mL). Calculate %RSD for migration time and peak area of the monomer.
- Intermediate Precision: Repeat the precision experiment on a different day, with a different analyst and a different capillary. Combine data for statistical analysis.
- Accuracy (by Spiking): Spike known amounts of a purified mAb fragment (e.g., 5%, 10%, 15%) into the mAb monomer standard. Calculate recovery: (Measured % - Baseline %) / Spiked % x 100.
- LOQ/LOD: Serially dilute an impurity (fragment/aggregate) standard. LOD: Concentration yielding S/N ~3. LOQ: Lowest concentration with %RSD <20% and S/N >10.
Data Analysis:
- Use instrument software (e.g., 32 Karat) to integrate peaks.
- Report % relative peak area for monomer, aggregate, and fragments.
- Compare all results against pre-defined acceptance criteria (e.g., Table 1).
Protocol 2: Analysis of Oligonucleotide Drug Substance Purity
Title: Purity and Identity Testing of Synthetic Oligonucleotide by CGE-LIF.
Objective: To determine the full-length purity and identity of a 20-mer synthetic oligonucleotide drug substance.
Methodology:
- Sample Preparation:
- Prepare a 0.1 mg/mL solution of the oligonucleotide in nuclease-free water.
- Prepare an ssDNA ladder solution per manufacturer's instructions.
- Combine 90 μL of sample (or ladder) with 10 μL of internal standard (e.g., 50 μM fluorescently-labeled marker) and 100 μL of sample buffer (containing formamide and EDTA).
Instrument Setup:
- Gel Matrix: Commercial dsDNA 1000 or similar gel matrix.
- Capillary: 100 μm ID, 30 cm total length (20 cm effective) coated capillary.
- Run Buffer: Commercial oligonucleotide separation buffer.
- Conditions: +15 kV for 25 minutes. Temperature: 30°C.
- Injection: Pressure injection at 0.5 psi for 20 seconds.
- Detection: LIF (ex/cm as per dye label, e.g., 488/520 nm).
Execution:
- Run the ssDNA ladder to create a calibration curve (log bp vs. migration time).
- Run the test sample in triplicate.
- Identify the main peak (full-length 20-mer) and failure sequence peaks (N-1, N-2, etc.).
Data Analysis:
- Calculate % full-length purity: (Area of main peak / Total area of all oligonucleotide peaks) x 100.
- Confirm identity: Migration time of the main peak should correspond to the 20-mer in the ladder (± 0.5 min).
- Report individual impurity peaks ≥ LOQ (typically 0.1%).
Visualizations
Title: ICH Q2(R2) Analytical Validation Workflow for CGE-LIF
Title: CGE-LIF Applications Across Biologics & Regulatory Links
The Scientist's Toolkit: Key Research Reagent Solutions
Table 4: Essential Materials for CGE-LIF Analysis of Biologics
| Item | Function & Description | Example Vendor/Kit |
|---|---|---|
| SDS-MW Analysis Kit | For size-based purity analysis of proteins under denaturing conditions. Contains gel matrix, run buffer, sample buffer, and standards. | Beckman Coulter (ProteomeLab), SCIEX |
| dsDNA 1000/5000 Kit | For sizing and purity analysis of oligonucleotides (0.1-1000 bp). Provides gel matrix, buffer, and internal standards. | Beckman Coulter (ProteomeLab) |
| Bare Fused Silica Capillaries | Standard capillaries for SDS-MW separations. Various internal diameters and lengths. | Beckman Coullet, Polymicro |
| Coated Capillaries (e.g., DB-1) | For oligonucleotide or native protein analyses to suppress electroosmotic flow (EOF) and analyte adhesion. | Beckman Coulter |
| Fluorescent Labels (for LIF) | Amine-reactive dyes (e.g., 5-FAM, Alexa Fluor 488) for labeling proteins or oligonucleotides if not intrinsically fluorescent. | Thermo Fisher, Sigma-Aldrich |
| Protein/ssDNA Ladders | Molecular weight/size standards essential for system suitability, identity confirmation, and migration time normalization. | Beckman Coulter, Thermo Fisher |
| Internal Standard (Fluorescent) | A consistently migrating fluorescent compound used to normalize injection variability and migration time (Critical for precision). | e.g., 50 μM Fluorescein |
| Stability-Indicating Stress Materials | For specificity studies: reagents for forced degradation (e.g., H2O2 for oxidation, heat for aggregation). | N/A (Lab prepared) |
Within the broader thesis of advancing Capillary Gel Electrophoresis with Laser-Induced Fluorescence detection (CGE-LIF) as a platform technology for biologics, this case study demonstrates its practical implementation as an orthogonal method. The thesis posits that CGE-LIF offers unparalleled sensitivity and resolution for analyzing critical quality attributes (CQAs) like size heterogeneity, charge variants, and glycosylation patterns. This application note validates its role in characterization and stability-indicating release testing, complementing traditional methods like SDS-PAGE and SEC-HPLC.
Application Note: Comparative Analysis of mAb Fragmentation
Objective: To assess the ability of CGE-LIF to quantify low-level fragmentation in a monoclonal antibody (mAb) drug product and compare its performance to traditional Size Exclusion Chromatography-High Performance Liquid Chromatography (SEC-HPLC).
Experimental Design: A stressed mAb sample (heat-treated at 45°C for 14 days) was analyzed alongside a control sample using both CGE-LIF and SEC-HPLC.
Key Research Reagent Solutions:
| Reagent/Material | Function in CGE-LIF |
|---|---|
| Fluorescent dye (e.g., 5-FAM SE) | Covalently labels primary amines (lysine residues) on proteins, enabling highly sensitive LIF detection. |
| Denaturing Gel Buffer (e.g., SDS-MW Gel Buffer) | Contains SDS to uniformly coat proteins with negative charge and linear polymers to act as a molecular sieve for size-based separation. |
| Coated Capillary (e.g., hydrophilic polymer-coated) | Minimizes protein adsorption to the capillary wall, ensuring reproducible migration and peak shape. |
| Size Ladder (Fluorescently labeled) | Provides internal migration references for accurate molecular weight estimation of sample components. |
Data Presentation:
Table 1: Quantification of mAb Fragments by CGE-LIF vs. SEC-HPLC
| Method | Parameter | Control Sample | Stressed Sample |
|---|---|---|---|
| CGE-LIF | Main Peak (%) | 98.7 ± 0.3 | 91.2 ± 0.5 |
| Heavy Chain (HC) Fragment (%) | 0.5 ± 0.1 | 4.1 ± 0.2 | |
| Light Chain (LC) Fragment (%) | 0.4 ± 0.1 | 3.5 ± 0.2 | |
| Low Molecular Weight (LMW) Species (%) | 0.4 ± 0.1 | 1.2 ± 0.1 | |
| Limit of Detection (LOD) | ~0.1% | ~0.1% | |
| SEC-HPLC | Main Monomer (%) | 99.1 ± 0.2 | 95.8 ± 0.3 |
| High Molecular Weight (HMW) Species (%) | 0.7 ± 0.1 | 1.5 ± 0.1 | |
| Fragment Species (%) | 0.2 ± 0.1 | 2.7 ± 0.2 | |
| Limit of Detection (LOD) | ~0.5% | ~0.5% |
Conclusion: CGE-LIF provided superior resolution of fragment species (distinguishing HC, LC, and other LMW fragments) and demonstrated approximately 5x lower LOD compared to SEC-HPLC, making it more suitable for detecting low-abundance degradants in release testing.
Detailed Experimental Protocols
Protocol 1: CGE-LIF Sample Preparation and Labeling for Purity Analysis
Principle: Proteins are denatured, reduced (if needed), and covalently labeled with a fluorophore for sensitive detection.
- Sample Dilution: Dilute the protein sample to a target concentration of 1 mg/mL in a neutral, amine-free buffer (e.g., 50 mM phosphate buffer, pH 7.5).
- Reduction (Optional, for detailed fragment mapping): Add 5 µL of 1M Dithiothreitol (DTT) to 95 µL of the diluted sample. Incubate at 70°C for 10 minutes.
- Labeling Reaction:
- Prepare a 10 mM stock of fluorescent dye (e.g., 5-FAM SE) in anhydrous DMSO.
- Add a 10-fold molar excess of dye stock to the protein sample. Vortex gently.
- Incubate the reaction in the dark at room temperature for 30 minutes.
- Quenching & Dilution: Stop the reaction by adding 1 µL of 1.5M hydroxylamine (pH 8.5). Mix and incubate for 15 minutes in the dark. Dilute the labeled sample 1:10 with deionized water prior to injection.
Protocol 2: CGE-LIF Instrumental Method for Size-Based Separation
Instrument: CGE-LIF system (e.g., PA 800 Plus or similar) with LIF detector (excitation: 488 nm, emission: 520 nm).
- Capillary Conditioning: Flush a coated capillary (50 µm ID, 30.2 cm total length, 20 cm effective length) with:
- Gel buffer for 5 min.
- Deionized water for 3 min.
- Gel buffer for 5 min.
- Sample Injection: Hydrodynamically inject the labeled sample at 5.0 kPa for 10 seconds.
- Separation: Apply a constant voltage of 15 kV (reverse polarity) for 30 minutes. Maintain capillary temperature at 25°C.
- Data Analysis: Use proprietary software (e.g., 32 Karat) to integrate peaks. Identify species by comparing migration times to a labeled molecular weight ladder. Report relative peak area percentages.
Visualized Workflows and Relationships
Title: Strategic Workflow for Orthogonal CGE-LIF Implementation
Title: CGE-LIF Sample Prep and Instrumental Analysis Workflow
Conclusion
Capillary Gel Electrophoresis with Laser-Induced Fluorescence detection (CGE-LIF) stands as an indispensable, high-resolution analytical tool in the modern biopharmaceutical toolkit. By combining the superior size-based separation of gel-filled capillaries with the exceptional sensitivity of LIF detection, it addresses critical needs in the characterization of complex biomolecules, from monoclonal antibodies to advanced gene therapies. Mastering its foundational principles, robust methodologies, and optimization strategies empowers researchers to generate high-quality data that drives development and ensures product quality. As therapeutic modalities continue to evolve in complexity, the role of CGE-LIF is poised to expand, particularly in the analysis of next-generation oligonucleotides, mRNA vaccines, and biosimilars. Future developments will likely focus on increased automation, integration with mass spectrometry, and harmonized data analysis platforms to further solidify its position as a gold-standard technique for purity and heterogeneity assessment in regulated environments.